Recommended by:

Top 20 UK science resources,
The Tutor Website
Recommended by:

Rated:

2010
Listed on the science, engineering and technology section of

'providing you with access to the very best Web resources for education and research, evaluated and selected by a network of subject specialists.'
(Please note that intute closed in July 2011)
Section 2: Inorganic
CHAPTER 13:
PERIODICITY IN PROPERTIES
OF THE ELEMENTS UP TO ARGON
NB This chapter has now been
updated to improve browser compatibility.
Please
use the 'send email' link at the top right hand corner of this page to
report any problems.
13.1. INTRODUCTION TO INORGANIC
13.1.1. THE KEY to inorganic (and organic) chemistry is electrostatic forces. The most likely reactions are those in which:
i) The forces of attraction are greater in the products than they are in the reactants (thermodynamic effect i.e. predicts position of equilibrium).
ii) However, forces in the surroundings must also be considered, as reflected by changes in overall entropy (chapter 12).
iii) The forces of attraction in any intermediate stage are not too much weaker than those in the reactants (kinetic effect i.e. predicts rate).
See also section 14.4.2.v.
The difficulty is constructing in your mind a picture of the changes which embraces all the electrostatic forces involved in the course of a reaction.
All these forces boil down to the attraction between positive protons and negative electrons. However, your task is to picture the various relative positions in which protons and electrons occur, and the relative strengths of the forces between them.
The forces range from the attraction of an element's nucleus for its own electrons to its attraction for the electrons of another element, from the attraction between oppositely charged ions to the attraction within molecules, and from strong individual bonds to those which are significant because of their quantity. STOP, to think of specific examples.
Moreover, reaching a state where the total electrostatic forces are greater than they were at the start of a reaction may involve a stage where negative charge is pulled a long way from positive charge. In such cases, reactant particles may have to collide with great speed to pass through that stage, and the reaction may therefore be impractically slow or require very high temperatures (sections 9.4. and 9.5.)
13.1.2. THE DOOR opened by an understanding of electrostatic forces is the periodic table. Opening this door reveals a pattern of periodic and group trends which organises the vast amount of information covered by inorganic chemistry.
The importance of the periodic table has already been discussed in chapters 2 and 4. In chapter 4 it was stated that inorganic facts should be learned only as examples of the two main patterns in the periodic table:
i) increasing effective nuclear charge across a period
ii) increasing size (number of shells) down a group.
The stunning fact is that most UK inorganic chemistry books hardly mention effective nuclear charge. They may attach some importance to the changes in size down a group, but effective nuclear charge does not even get a mention in the index of many UK text books at this level. The concept fares better in US text books and certainly gets a mention on Wikipedia.
If you want to spend a long time learning, forgetting, and re-learning a tedious (but intellectually undemanding) list of facts, ignore the patterns in the periodic table. If you think it is worth understanding and remembering what you read, by working harder, (though for a shorter time) read on.
13.2. EFFECTIVE NUCLEAR CHARGE
Effective nuclear charge is a measure of the attraction between a nucleus and its outer shell electrons. Clearly, it depends on the number of protons in the nucleus (i.e. actual nuclear charge), but it also depends on the number of inner shells which screen the outer shell from the effect of the nucleus.
The screening effect of inner shells varies. First, the smallest, inner, shells are the most effective screeners. Second, within a particular shell/energy level screening decreases in the order s-orbtals > p-orbitals > d-orbitals > f-orbitals. This is because the orbitals become less compact, more spread out, and more directional, in this order.
13.3. BONDING AND GROSS PHYSICAL PROPERTIES
Changes in type of bonding across a period have already been dealt with in sections 4.4. and 4.6.). From these changes, changes in gross physical properties such as state and appearance can also be predicted. The changes in oxidation state across a period have also already been discussed (section 4.7.).
13.4. ATOMIC RADIUS
13.4.1. The changes in atomic radius down a group and across a period, have already been mentioned in chapter 4. However, there are three measures of atomic radius, and these are discussed here.
13.4.2. Covalent radius is half the distance beteen the centres of two atoms in a diatomic molecule of an element. The distance is determined by X-ray diffraction or electron diffraction (section 5.2.2.).
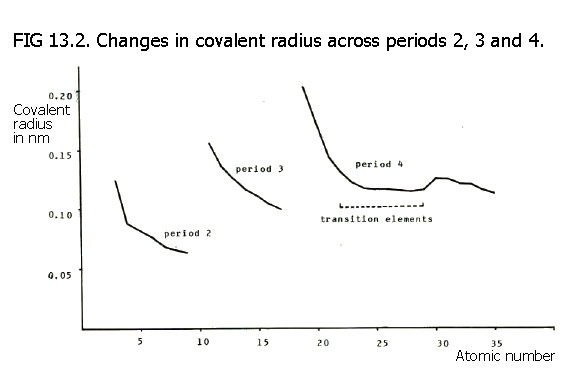
Covalent radius decreases across a period owing to the increase in effective nuclear charge i.e. the increase in number of protons attracts the bonding electrons more strongly and pulls the atoms closer together. This is because there is no increase in number of electron shells across the periods.
13.4.3. Metallic radius is half the distance between the centres of two adjacent atoms in a metallic lattice. The distance is again measured by diffraction tecniques.
Metallic radius decreases across a period for the same reasons that covalent radius decreases. Again the reasons can be summarised by the phrase "increase in effective nuclear charge". The increase in effective nuclear charge across a period means that the ions within the metallic bonding cloud of electrons are pulled closer together by their increasing attraction for the electrons in that cloud.
Metallic radius is generally greater than the covalent radius for the same element, because the bonding electrons in the metallic bond are more diffuse and therefore the attraction between the nuclei and the bonding electrons is weaker.
13.4.4. Van der Waals radius is half the distance between the centres of two colliding atoms which are not otherwise bonded to each other.
It, too, decreases
across a period owing to increasing effective nuclear charge. Moreover,
as expected, van der Waals radius for a particular element is greater
than either its metallic radius or its covalent radius.
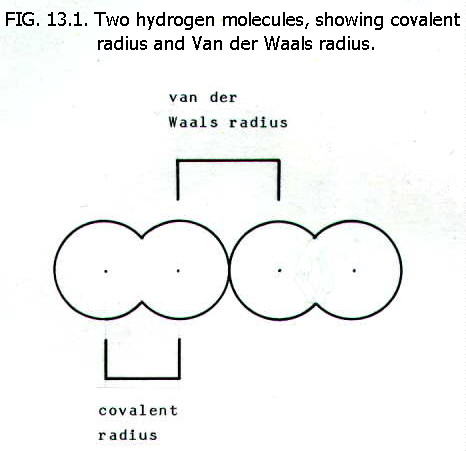
13.4.5. To sum up,
the three measures of atomic radius decrease in the order van der Waals
radius > metallic radius > covalent radius, for a
particular element, though it may not be possible to make each of these
measurements for a given element.
Whatever measure is
used, atomic radius decreases across a period. Note also, that the
atomic radii in period 3 will generally be greater than those in period
2, as predictable from the additional electron shell in the shell
model. In fact, it is necessary to travel along the third period as far
as silicon (group IV) before finding an element whose covalent radius
is smaller than that of lithium.
13.5. IONIC RADIUS
13.5.1. Ionic radius is that part of the distance between the centres of two adjacent ions in an ionic lattice which is due to the ion in question.
Diffraction techniques are used to measure distances between nuclei, and then other factors are used to estimate the part of the distance due to each ion. These factors include the atomic radius of the parent atom(s), and the accumulation of data from several ionic lattices for each ion.
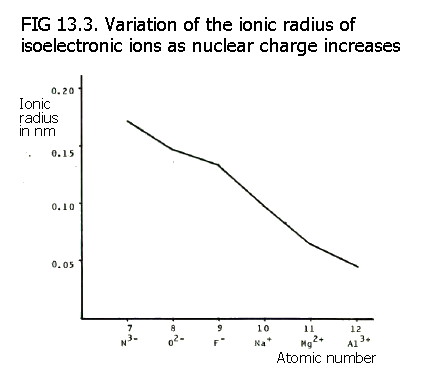
13.5.2. Trends and changes across a period are perhaps a little meaningless due to the changing nature of the ions. In the second period there is a useful comparison of the ions Li+, Be2+, and B3+, because they all have the same electron configuration, 1s2. They are said to be isoelectronic. Apart from the increasing gross charge on the ions, the only change along the series is the increasing number of protons. Thus size decreases.
Positive ions are, of course, smaller than their parent atoms because they have completely lost a shell. Negative ions, by contrast, are bigger than their parent atoms because they have more electrons which need to be held in by the same nucleus. Thus the N3-, O2-, and F- ions are all larger than the positive ions formed by the elements at the start of the second period. Apart from the more esoteric arguments about loss or gain of electrons, the negative ions have one more shell of electrons than the positive ions.
However, the negative ions listed in the last paragraph decrease in size in the order shown. They all have the same electron configuration 1s2. 2s2. 2p6, but the nuclear charge (and therefore effective nuclear charge) increases along the N3-, O2-, F- series.
N3-,
O2-, and F- are also
isoelectronic with Na+, Mg2+,
and Al3+. These positive ions have even greater
nuclear charge. They are therefore smaller than the negative ions
formed by the elements at the end of the previous period. As with the
ions at the beginning of period 2, the size of these positive ions
decreses from Na+ to Al3+.
At the other end of the third
period, only sulphur and chlorine form simple negative ions. The outer
shell of phosphorus is further from the nucleus than that of nitrogen,
so it is unable to attract 3 electrons into the shell and form a P3-
ion.
O2- and Cl- are bigger than the positive ions at the start of the period because they have gained electrons and have one more shell of electrons than the positive ions. In addition, these negative ions are bigger than the corresponding ions in the previous period, because they have an extra shell.
13.6. IONISATION ENERGY
13.6.1. Ionisation
energy is described in section 6.6.2.iii. It is measured
using data from atomic spectra (FIG. 2.3.) or using the electron impact
method.
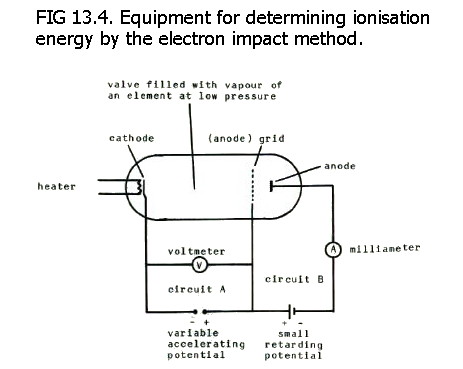
The valve is filled with
monatomic gas of the element at low pressure.
Electrons are attracted from the heated cathode towards the anode grid and flow round circuit A. As the potential difference in circuit A is increased, more and more electrons pass through the grid, overcome the retarding potential in circuit B, reach the (so-called) anode, and flow round circuit A+B. The current measured by the milliameter therefore increases.
However, as the *potential difference in circuit A is increased yet further, some of the electrons have inelastic collisions with the atomised element. These electrons do not reach the "anode", and the milliameter records a decrease in current. (*The voltmeter measures the potential difference applied across the cathode and the grid.)
These inelastic collisions excite electrons in the atom to higher energy levels, and the peak current recorded on the ammeter corresponds with an excitation potential recorded by the voltmeter.
Increasing the potential difference even further, again increases the passage of electrons through the grid. Further excitation potentials may be recorded, but eventually, collisions occur with sufficient energy to completely remove electrons from the atomised element. Electrons which collide and cause ionisation do not reach the anode.
Thus another peak current is measured by the ammeter, and in this case it corresponds with the first ionisation potential recorded by the voltmeter. Ionisation potential can then be converted into first ionisation energy because the energy attained by one mole of electrons accelerated across a potential difference of 1 volt = 96.3 kJ.
Continuing the increase in potential difference in circuit A allows the measurement of further ionisation energies.
13.6.2. Changes
in first ionisation energy for the elements from hydrogen to
argon are shown in FIG. 13.5. below.
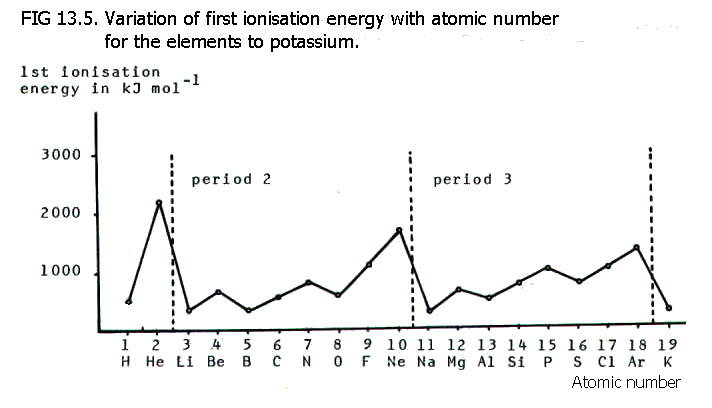
There is a general increase in
ionisation energy across a period, as can be predicted from the
increase in effective nuclear charge across a period. The increase in
effective nuclear charge means that the outer electrons are held more
tightly, and more energy is needed to remove them.
The big drop in ionisation energy from one period to the next is also predictable. As a new period is started, the outer electron is in a shell further from the attraction of the nucleus. Far less energy is therefore needed to remove it.
The elements in the third period generally have lower ionisation energies than the corresponding elements in the previous period. This is again predictable because the elements in the third period have an extra shell.
The apparent irregularities in the two longer periods are also predictable. The first p-electron in each period (in boron and aluminium) is more easily removed than the previous s-electrons, because the pair of s-electrons quite effectively screens the p-orbitals in the same shell/energy level (section 13.2.)
The first paired p-electrons in oxygen and sulphur are also relatively easy to remove. This is a characteristic of the uneven electron distribution (and therefore charge distribution) in oxygen and sulphur compared with that in half filled p-orbitals. This enables prediction of the falls in first ionisation energy between nitrogen/oxygen, and phosphorus/sulphur.
Note also that half-filled and completely filled p-orbitals have an even charge distribution and are often referred to as having extra stability as a result of this.
13.6.3. The first few ionisation energies of a partciular element give a great deal of information about the element.
In particular, since the outer shell electrons are furthest from, and most screened from, the nucleus, they are much easier to remove than subsequent electrons and have much lower ionisation energies.
There is therefore a
jump in ionisation energy as electrons are removed from the next shell,
and the number of low ionisation energies enables prediction of the
number of outer electrons, and hence group number etc.
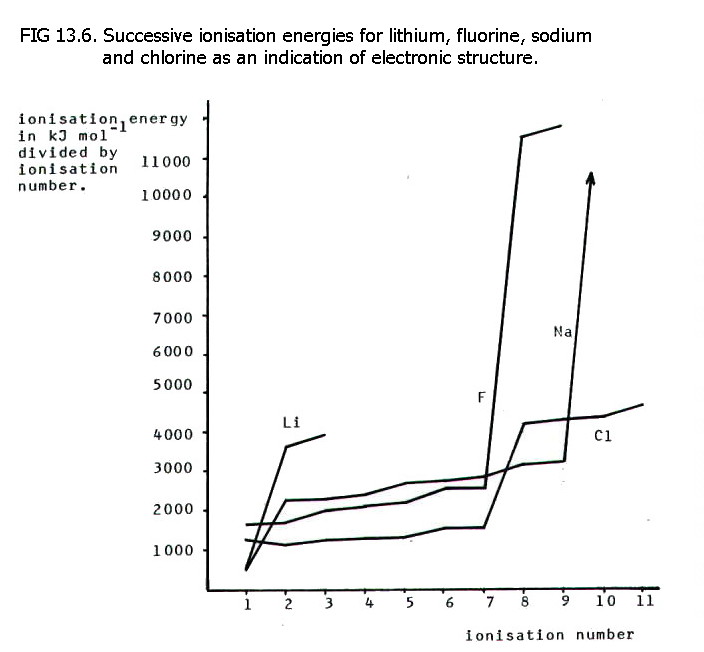
Note also, that the relative
magnitudes of the first ionisation energies indicate the relative
positions of lithium and sodium, and of fluorine and chlorine. The total
number of ionisation energies of lithium also gives a clear indication
of position in the periodic table!
13.7. ELECTRON AFFINITY
13.7.1. Electron affinity has already been described in section 6.6.2.iv. Its direct measurement is difficult, so it is usually calculated using other data in combination with Hess's law.
Since effective nuclear charge increases across a period, electrons will be more strongly attracted into the outer shell as the period is crossed. Thus less energy will be required to add an electron as the period is crossed and electron affinity will become less positive and more negative i.e. since most first electron affinities are negative, they will increase numerically across the period.
Some idiosyncrasies include: Nitrogen has a first electron affinity of 0kJmol-1. This is because adding an electron makes the charge distribution in the p-orbitals uneven, compared with an even charge distribution in the half-filled orbitals.
Fluorine's first electron affinity is less negative than chlorine's. This is because the incoming electron must fit in with seven electrons that are already in a very small shell. A similar situation occurs with oxygen and sulphur.
13.7.2. Second electron affinity is always endothermic. This is because a second electron is always being added to an already negative ion, thus it is electrostatically repelled.
13.8. MELTING POINT AND BOILING POINT
13.8.1. Changes in m.p. and b.p. across a period are obviously very dependent on type of bonding (section 4.6.). This is because melting and boiling involve increasing temperature until the random motion of particles is vigorous enough to overcome the bonding between them. The stronger the bonds, the more vigorous must be the motion, and the higher must be the temperature.
As described in section 4.4.3., the relatively low effective nuclear charge in elements on the left of the table results in metallic bonding. Since it is the metallic bonds which break when metals melt or boil, these metals have fairly low melting points and boiling points.
However, the increase in effective nuclear charge across the periods causes an increase in the strength of metallic bonding. This, in turn, results in an increase in melting point and boiling point.
The extra shell (and therefore extra distance between the nucleus and the bonding electrons) in the third period, makes the bonding generally weaker than that in the second period. Melting point and boiling point are therefore correspondingly lower.
Thus, in the second period m.p. and b.p. reach a peak at boron and carbon, where the structure has essentially become giant covalent rather than metallic. Carbon's m.p. is considerably higher than boron's but its b.p. is fractionally lower than that of boron. However, in the third period, the giant molecular structure does not exist until silicon. Both m.p. and b.p. reach a clear peak at silicon in the third period.
Note that it is not the changeover from metallic to giant molecular structure which itself causes the increase in m.p. and b.p. These are parallel changes which all result from the increased attraction of the nuclei for the bonding electrons i.e. from the increased effective nuclear charge.
|
TABLE 13.1. Melting and boiling
points (in °C) of the elements to argon |
||||||||
| element |
Li |
Be |
B |
C |
N |
O |
F |
Ne |
|
Melting point |
181 |
1283 |
2000 |
3800 |
-210 |
-219 |
-219 |
-248 |
|
Boiling point |
1331 |
2477 |
3900 |
3800 |
-196 |
-183 |
-188 |
-246 |
| element |
Na |
Mg |
Al |
Si |
P |
S |
Cl |
Ar |
|
Melting point |
98 |
650 |
659 |
1410 |
....44w **597r |
*96rh 119m |
-101 |
-189 |
|
Boiling point |
890 |
1117 |
2447 |
2680 |
281w †431r |
445m |
-34 |
-186 |
| *transition to monoclinic, **
at 143 atmospheres pressure, † sublimes w = white phosphorus, r = red phosphorus rh = rhombic sulphur, m = monoclinic sulphur |
||||||||
In both periods, simple covalent structures with low melting and boiling points take over from group V onwards. The low transition temperatures represent the totally different process that takes place when simple covalent structures melt or boil. It is no longer the main bonding which breaks, but weak van der Waals forces between molecules.
In other words the arguments are different here. The increased effective nuclear charge causes a change in type of bonding which in turn causes a change in m.p. and b.p. The changes are not simply parallel in this case.
In the second period, nitrogen, oxygen, and fluorine are all diatomic gases with very similar melting points (between -210 and -220 °C) and boiling points (between -180 and -200°C).
The extra shell in the third period makes the transition to discrete diatomic molecules "slower". Phosphorus and sulphur still have fairly large molecules (section 4.6.3.). The van der Waals forces between moleucules are quite strong and although melting points and boiling points are much lower than the previous elements in the period, they are much higher than those of nitrogen and oxygen. In fact, both phosphorus and sulphur are solids.
Even chlorine has considerably higher melting and boiling points than fluorine, despite existing as a discrete diatomic molecule. This is because the extra shell makes the outer electrons further from the attractive influence of the nucleus. The types of instantaneous polarisation which give rise to van der Waals forces therefore occur more readily. This means the van der Waals forces will be stronger and will require higher temperatures before breaking.
Neon and argon have extremely low melting points because they exist as single atoms with very weak van der Waals forces between them. The boilng points hardly differ from the melting points because random atomic motion sufficient to break the solid structure is virtually sufficient to break all the van der Waals bondsing.
The reasons for the higher melting and boiling points of argon compared with neon are similar to those accounting for the differences between chlorine and fluorine.
13.9. METALLIC VS. NON-METALLIC BEHAVIOUR
13.9.1. Even changes in chemical behaviour can be predicted from the results of increasing effective nuclear charge.
FIG. 13.7. Division between metals and non-metals in the second and third periods

For example, the classification into metals and non-metals, shown in FIG. 13.7., invloves more than a quick glance at an element to see if it is a shiny solid. The type of bonding in each case has already been described in sections 4.4.3. and 4.4.4., but there is also chemical behaviour which is characteristic of metals and of non-metals.
13.9.2. Electropositivity and electronegativity. Metals are electropositive; they tend to lose electrons and form positive ions. Non-metals are electronegative; they tend to gain electrons and form negative ions.
We have already predicted a change from electropositive to electronegative behaviour across a period: on the left hand side of the table, effective nuclear charge is low, allowing elements to lose electrons; on the right hand side, effective nuclear charge is high, enabling elements to gain electrons (section 4.3.3.). In other words there is a change from metallic to non-metallic behaviour, which parallels the change in bonding (section 4.6.3.).
13.9.3. With water, for example, elements on the left hand side of the table tend to form aqueous cations, and elements on the right hand side tend to form aqueous anions:
E.g...
2Na(m)..
+..
2H2O(l)......![]() ...... 2Na+(aq)..
+..
2-OH(aq).. +..
H2(g)
...... 2Na+(aq)..
+..
2-OH(aq).. +..
H2(g)
.........Cl2(g)...
+..
H2O(l)........![]() ...... H+(aq)..
+..
Cl-(aq).. +..
HOCl(aq)
...... H+(aq)..
+..
Cl-(aq).. +..
HOCl(aq)
........................[HOCl(aq)...... ...... H+(aq)..
+..
-OCl(aq)]
...... H+(aq)..
+..
-OCl(aq)]
13.9.4. Acid-base behaviour. The equations in the last section show another important distinction between metals and non-metals.
Metals tend to dissolve in water and produce alkaline solutions,
whereas
non-metals tend to produce acidic solutions.
Moreover, remember that this is predictable as a direct consequence of the difference in effective nuclear charge.
In both periods, there is a transition point from basic to acidic behaviour. Since the elements in the third period have an extra shell, it can be predicted that the transition will occur later in this period. This is because the extra shell in the third period elements is further from the nucleus and electrons are therefore less readily attracted into the shell.
Thus, the transition occurs at beryllium in period 2 and at aluminium in period 3. Both berylium and aluminium are quite insoluble in water though both can be encouraged to show basic behaviour by dissolving in acid, or acidic behaviour by dissolving in alkali. The ability to show both basic and acidic behaviour is known as amphoteric behaviour.
..
Be(s)..
+..
2H+(aq).. .. Be2+(aq)..
+..
H2(g)
.......................................................................................
Basic behaviour
. 2Al(s)..
+..
6H+(aq).. .. 2Al3+(aq)..
+..
3H2(g)
Be(s).
+.
2-OH(aq). +.
2H2O(l).. .. 2Na+(aq).
+.
[Be(OH)4]2-(aq).
+.
H2(g)
......................................................................................
Acidic behaviour
2Al(s). +. 2-OH(aq). +. 6H2O(l).. .. 2Na+(aq). +. 2[Al(OH)4]-(aq). +. 3H2(g)
The behaviour is actually somewhat more complex than this, as shown by the scheme below for aluminium.
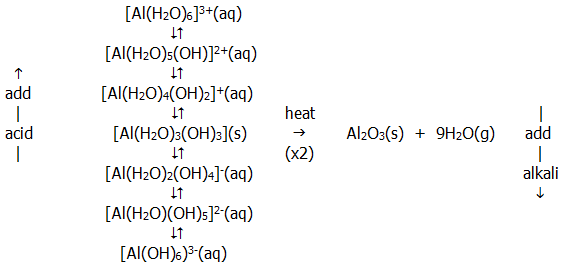
Note that the similarity between beryllium and aluminium is sometimes described as an example of a diagonal relationship. It results from the extra shell in aluminium counteracting the higher effective nuclear charge that is expected from its position nearer the right hand side of the periodic table.
Acid-base behaviour is discussed in more detail under oxides and hydroxides in the next chapter (section 14.5.2.).
13.10. QUESTIONS
1) List as many examples as possible of the forces described in paragraph 4 of section 13.1.1.
2) Explain how section 13.1. fits in with the ideas discussed in chapter 12.
3) Describe clearly the link between increasing effective nuclear charge across a period and the changes in van der Waals radius.
4) Explain why measurements of atomic radius for a particular element decrease in the order: van der Waals radius > metallic radius > covalent radius. Are all these types of measurement equally valid for every element? How can a valid comparison be made between the atomic radius of sodium and that of chlorine?
5) Explain why the difference in melting point and boiling point for metals is greater than it is for elements with a giant molecular structure.
6) Phosphorus and sulphur have higher melting points than might be expected. Their boiling points are not much higher than their melting points. Discuss the validity of these statements and describe how the melting and boiling points give information about the bonds broken during both melting and boiling.
7) Using a data book,
construct a table of  Hfusion
and Hvaporisation for the
elements litium to argon. Follow the layout in table 13.1. Explain the
changes across both periods in terms of i) the increase in effective
nuclear charge across the periods and ii) the extra shell in period 3.
Compare and contrast the changes across the periods with the changes in
melting point and boiling point, offering explanations for similarities
and for differences.
Hfusion
and Hvaporisation for the
elements litium to argon. Follow the layout in table 13.1. Explain the
changes across both periods in terms of i) the increase in effective
nuclear charge across the periods and ii) the extra shell in period 3.
Compare and contrast the changes across the periods with the changes in
melting point and boiling point, offering explanations for similarities
and for differences.
8) The pH of aqueous solutions of elements in the third period changes as the period is crossed. Explain how these changes are directly related to the changes in effective nuclear charge across the period.
9) Explain which of the schemes below best summarises the relationships for sodium between: effective nuclear charge, electropositive behaviour, metallic bonding, classification as a metal, and basic behaviour:

Unless otherwise stated, all materials in this web version of chapter 13 are © 2007 Adrian Faiers MA (Oxon) MCIPR

What 's the connection between a dozen eggs and
a garden mole?

Answer: Not a lot, really, but see Chapter 1