Recommended by:
Top
20 UK science resources,
The Tutor Website
Recommended by:

Rated:

2010
Listed on the science,
engineering
and technology section of 
'providing you with access
to the very best Web resources for education and research, evaluated
and selected by a network of subject specialists.'
(Please note that intute closed in July 2011)
Section 2: Inorganic
CHAPTER 16: GROUP
VII, THE HALOGENS
NB This chapter has now been
updated to improve browser compatibility.
Please
use the 'send email' link at the top right hand corner of this page to
report any problems.
16.1. INTRODUCTION
The chemistry of group VII is dominated by the high effective nuclear charge on the right of the periodic table. Just as the low effective nuclear charge in groups I and II lead to positive ion formation, so the high effective nuclear charge in group VII causes the elements to gain electrons and form negative ions (section 4.4.2.). As predictable from section 4.7.5. they gain one electron and form 1- ions.
But the high effective nuclear charge creates another possibility. Covalent bond formation was not a possibility with any but the most electronegative elements in groups I and II, because covalent bonds require nuclei to maintain control over bonding electrons. However, the high effective nuclear charge in group VII allows just this behaviour when a halogen is bonded to an element which is itself reasonably electronegative.
Moreover, a covalent bond between two highly electronegative elements will clearly be strong. Covalent bond formation is no less a characteristic of high effective nuclear charge than is negative ion formation, though negative ion formation is the more unique.
Down the group, the increasing number of shells leads to a decrease in strength with which the outer electrons are held, and with which the further electron is attracted into the outer shell. In other words, there is a decrease in electronegativity. Iodine even shows some of the characteristics of an electropositive element: it is a solid with a metallic lustre and even forms an I+ ion as a transition state in some reactions. I+ probably also exists in solutions of ICl in concentrated sulphuric acid.
A more typical example of the decreasing electronegativity is the decreasing ionic character of the aluminium halides. Aluminium fluoride is ionic, aluminium chloride has a high degree of covalent character, and the bromide and iodide show even more covalent characteristics like lower melting points, and solubility in organic solvents.
In this chapter we shall not discuss changes in such an extensive list of properties as we did for groups I and II. Properties such as atomic and ionic radius, melting point, boiling point etc. largely follow expected patterns. We shall discuss changes in properties selected for their particular interest. In addition, the anomalous behaviour of fluorine and its compounds will come in for special mention.
16.2. THE ELEMENTS
16.2.1. Changes in physical state down group VII are a good reflection of the increasing metallic character:
Fluorine:...... pale (greenish) yellow gas (b.p. -188 °C)
Chlorine:......(pale) yellowish green gas (b.p. -36 °C)
Bromine:..... reddish brown liquid (b.p. 58 °C)
Iodine:........ (violet) black solid
with metallic lustre (b.p. 183 °C
- normally
sublimes to give violet
gas)
Given how
electronegative iodine is, it may seem fanciful to say that the
transition down the group from gas ![]() liquid
liquid ![]() solid, and the increase
in colour (not to mention reflective properties) represent increasing
metallic character. However, the properties themselves reflect a
decreasing distinction between the intramolecular
covalent bonds which are becoming weaker (section 13.2.2), and the intermolecular
van der Waals bonds which are becoming stronger.
solid, and the increase
in colour (not to mention reflective properties) represent increasing
metallic character. However, the properties themselves reflect a
decreasing distinction between the intramolecular
covalent bonds which are becoming weaker (section 13.2.2), and the intermolecular
van der Waals bonds which are becoming stronger.
The van der Waals bonds become stronger down the group because the outer shell is further from the nucleus. This means that the instantaneous dipoles which give rise to van der Waals bonds become more marked, and less short-lived. Thus, the position of the outer electrons becomes less well-defined or, in other words, the decreasing distinction between the covalent and van der Waals bonding represents increasing metallic character in the bonding (section 4.6.3.).
16.2.2. i) Covalent bond strength decreases down group VII as the two nuclei become further from the covalent bonding electrons and therfore less strongly attracted to them. This is reflected in the values of the bond enthalpies (or atomisation enthalpies), which decrease down the group.
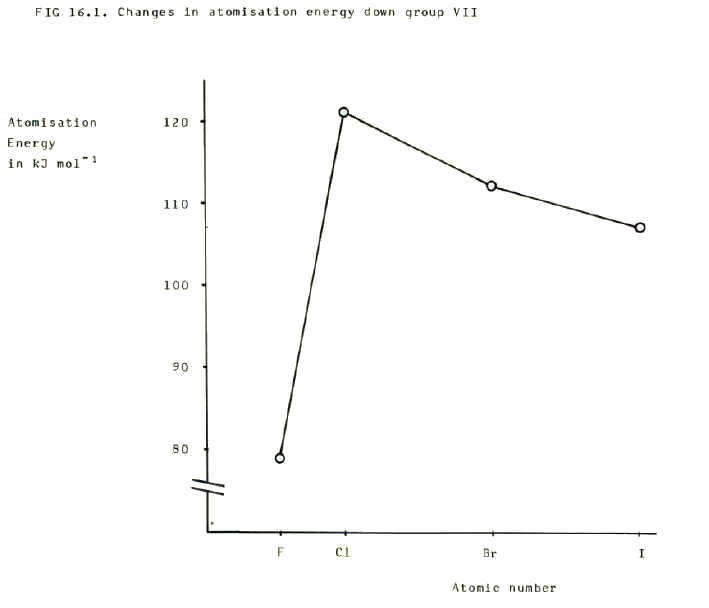
ii) However, fluorine's bond is weaker than chlorine's, and this is a function of its very small size. It is true that the nuclei should be very strongly attracted to the bonding electrons, but also the non-bonding electrons in one atom are very close to the non-bonding electrons in the other atom. This leads to repulsive forces between the atoms and weakening of the bond. The relative weakness of the bond is reflected in the bond enthalpy.
16.2.3. i) The ability to gain an electron decreases down the group and this is reflected in a numerical decrease in the electron affinities (they becomes less negative).
Again this is predictable from the increasing number of shells. As the outer shell becomes further from the attractive power of the nucleus, it becomes less able to gain an electron.
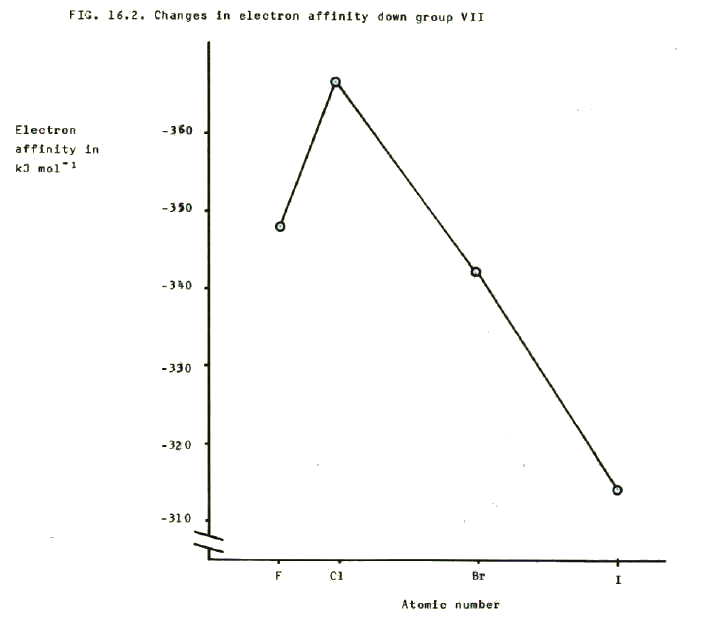
ii) However, fluorine is less able to gain an extra electron than chlorine. This again is a property of fluorine's particularly small size. The incoming electron is repelled to some extent by the electrons already present in the atom's very small outer shell. The fact is reflected in the value of fluorine's electron affinity which is numerically lower (less negative) than chlorine's.
16.2.4. i) The
force with which halide ions attract water molecules
(hydration) decreases down the group. This is because the increase in
size means a decrease in surface charge density of the ions and hence
decrease in electrostatic attraction for the 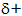 hydrogen atoms of water
molecules. The fact is reflected as a numerical decrease in the
(negative) values of the hydration energies.
hydrogen atoms of water
molecules. The fact is reflected as a numerical decrease in the
(negative) values of the hydration energies.
ii) Fluoride ions attract water molecules even more strongly than expected owing to their particularly small size and particularly high surface charge density. The hydration energy of fluoride ions has a particularly high negative value.
16.2.5. Oxidising power of halogens in water.
i) Changes down the group in the oxidising power of halogens in water, can be predicted using the predictions in the last three sections (16.2.3t5.).
When a halogen acts as an oxidising agent in water, it undergoes the following reduction:
X2(s,l,or
g)..... +..... 2e-........![]() ........2X-(aq)
........2X-(aq)
In other words it has to undergo vapourisation (when relevant), atomisation, gain of an electron, and hydration, though it should not be imagined that this represents a real sequence of events (c.f. section 15.2.7.).
Even with iodine, the electrostatic forces which are overcome during vapourisation are insignificant compared with the other electrostatic forces operating in the reaction.
So, down the group:
a) The halogens show an increasing ability to atomise which tends to increase oxidising power.
b) The halogens show a decreasing ability to gain electrons which tends to decrease oxidising power.
c) The halogens show a decreasing attraction for water molecules (hydration) which tends to decrease oxidising power.
Overall, effects b) and c) outweigh a), and there is decrease in oxidising power down the group.
This prediction can be tested by looking up values for the relevant enthalpy changes, and using enthalpy diagrams such as the one in FIG 16.4. to analyse the data.
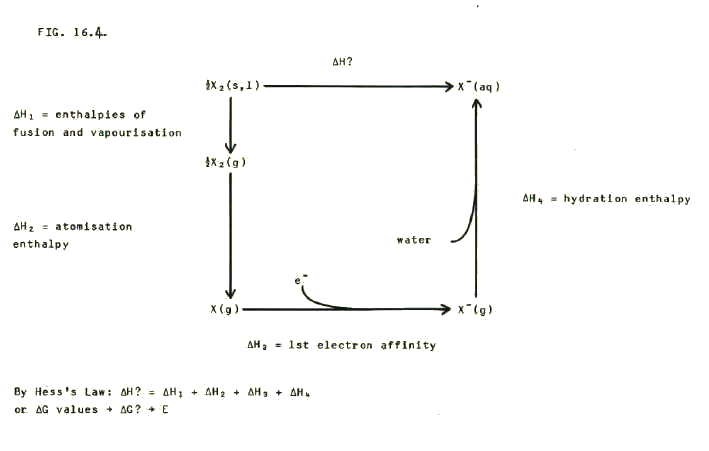
The decrease in oxidising power is also reflected as a decrease in the redox potentials, as shown in FIG. 16.5.
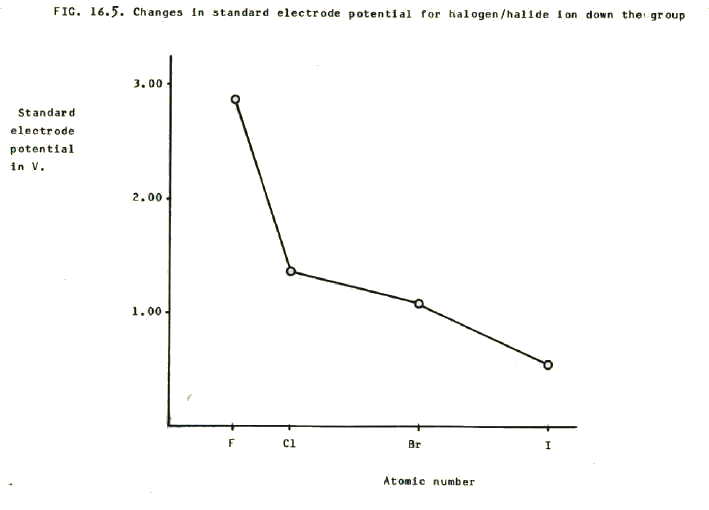
Note that the redox potentials include both enthalpy and entropy in the calculations (section 12.5.2.).
ii) Fluorine is an even stronger oxidising agent than expected. This can be predicted from its weaker than expected bonding, and its particularly strong attraction for water molecules, which outweigh its lower than expected ability to attract electrons.
Not only is fluorine a thermodynamically strong oxidising agent, it is also a very fast oxidising agent. This is predictable from the particularly weak F-F bond. When fluorine acts as an oxidising agent (not soley in aqueous solution) the F-F bond must be broken at some stage. This is probably the rate-determining process, and the ease with which is occurs gives a fast overall rate.
Even the other halogens are relatively fast oxidising agents compared, say, with the more electronegative element, oxygen. This supports the bond strength argument. Oxygen has a strong double bond and high temperatures are needed before it oxidises at a reasonable rate. Nitrogen is not even thought of as an oxidising agent, and yet its effective nuclear charge, as indicated by the Pauling electronegativity value (section 14.4.2.vi.), is about the same as chlorine's.
iii) Displacement reactions are a good example of the relative oxidising powers of halogens. Halogens are displaced from aqueous solutions of their ions by halogens higher in the group. E.g. bromine is displaced by chlorine from an aqueous solution containing bromide ions. The chlorine is reduced to aqueous chloride ions during the process:
Cl2(g)..... +.....
2Br-(aq)........  ........2Cl-(aq)..... +.....
Br2(aq)
........2Cl-(aq)..... +.....
Br2(aq)
iv) The relative abilities of halogens to oxidise water are discussed in section 16.3.3.
v) Even the preparation of hydrogen halides is determined by the relative oxidising powers of the halogens.
A simple method used to prepare hydrogen chloride or hydrogen fluoride is to heat an appropriate salt (sodium fluoride or sodium chloride) with concentrated sulphuric acid:
E.g....NaCl(s)... +...
H2SO4(l)..... .....HCl(g)...
+...
NaHSO4(s)
However, concentrated sulphuric acid is also an oxidising agent and it is powerful enough to oxidise bromide and iodide ions to the respective halogens. The method produces hydrogen halide contaminated with halogen and sulphur dioxide:
E.g...2HBr(g)...+..
H2SO4(l)... .. .Br2(g).. +..
2H2O(g)..
+.. SO2(g)
In contrast, concentrated phosphoric acid is not a powerful enough oxidising agent to oxidise bromide and iodide ions. Thus it can be used as an alternative in the preparation of all the hydrogen halides from appropriate salts.
vi) The action of other oxidising agents on halides also reflects the pattern.
Even the strongest laboratory oxidising agents are unable to oxidise hydrogen fluoride. Only the most powerful (e.g. MnO2 and KMnO4) oxidise HCl to chlorine gas, a reaction used to prepare chlorine: concentrated hydrochloric acid is dripped onto solid KMnO4. Chlorine prepared in this way may then be used to prepare chlorides (section 14.6.).
Hydrogen chloride can be oxidised by air, but only in the presence of a heated catalyst such as copper (I) chloride:
4HCl(g)..... +.....
4O2(g)........ ........2Cl2(g)..... +.....
2H2O(g)
As already seen in section iv above, bromides are far more readily oxidised even by moderately powerful oxidising agents. Crystals of white bromides slowly turn yellow in air as bromide ions are oxidised to bromine, and aqueous solutions slowly turn orange for the same reason.
Hydrogen iodide is a powerful reducing agent:
E.g.... 2I-(aq)... +...
2Fe3+(aq)..... .....I2(aq)....+....2Fe2+(aq)
Moreover, when it is oxidised to iodine by concentrated sulphuric acid (section v above) some of the sulphur is reduced to an oxidation state of -II and hydrogen sulphide is produced. Crystals of white iodides are not only more likely than those of bromides to turn yellow in air, but they do so more rapidly. The solutions turn brown. This is because the iodine produced by aerial oxidation forms a brown complex with the iodide ions still present:
I2(aq)..... +.....
I-(aq)................I3-(aq)
....................................... ...... brown
This colour change, in either direction, is such a significant part of practical chemistry, it seems to deserve more than this brief mention. However, there is an even sharper colour change which is used in quantitative analysis.
Just as the I3- colour has reached pale straw in titrations of I2/I- solutions with thiosulphate ions, a few drops of aqueous starch are added. This produces a deep blue complex (section 25.3.5.ii). It is the disappearance of this blue colour which is taken as the end point in the titration:
I2/starch(aq).. +..
2S2O32-(aq)..... .....2I-(aq).. +..
S4O62-(aq).. +..
starch(aq)
blue/black
16.3. THE COMPOUNDS
16.3.1. i) Hydrogen-halide bond strength decreases down the group. This can be predicted because the increasing number of shells means that the bonding electrons become further from the halogen nucleus as the group is descended. The electrostatic attraction which holds the molecule together therefore decreases. The changes are reflected as a decrease in bond dissociation enthalpy down the group.
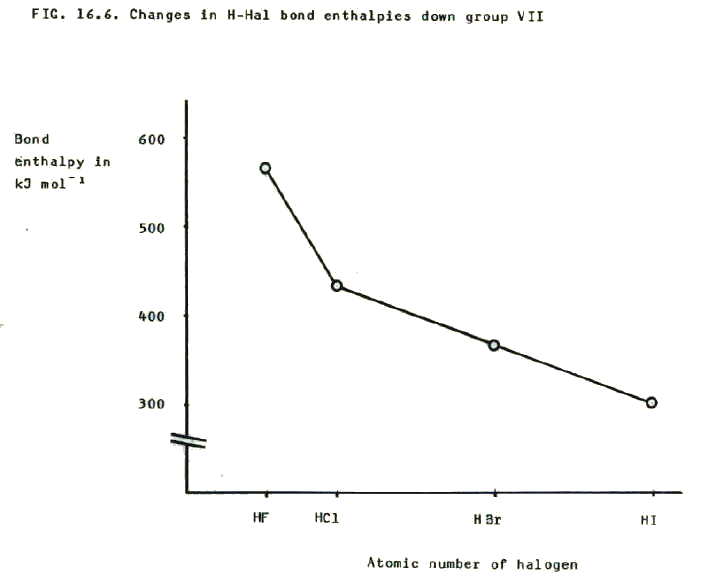
ii) The H-F bond is particularly strong because the fluorine atom is so small that electrostatic attraction of both nuclei to the bonding pair of electrons is particularly strong. Unlike the F-F bond (section 16.2.2.ii.) there is no weakening of the bond due to repulsion between non-bonding electrons: hydrogen does not have any. The strength of the H-F bond is reflected in the high H-F bond dissociation enthalpy.
iii) But boiling point and melting point of the hydrogen halides increases from HCl to HI because it is, of course, not the covalent bonds which break when the hydrogen halides melt or boil; it is the weak intermolecular forces and the increasing size of the halogen atoms results in increasing total strength of Van der Waals forces. The importance of the increasing Van der Waals forces may be a little surprising given H-Hal molecules are polarised and you might expect the deciding factor to be weakening permanent dipole - permanent dipole bonding down the group. However, the difference in electronegativity between hydrogen and the halogen from Cl to I is not that significant (0.9 reducing to 0.4). By contrast, the difference in HF is 1.9 and the resuting hydrogen bonding in HF results in its anomolous high melting point and boiling points, HF being a liquid at room tempoerature.
16.3.2. i) Relative acid strengths of the hydrogen halides can be predicted from a combination of the predictions in sections 16.3.1., 16.2.3., and 16.2.4. This is because the acid strength is a function of the dissociation of hydrogen halide in water. This in turn involves bond dissociation, gain of an electron by the halogen, and hydration of the halide ion. It also involves ionisation of hydrogen and hydration of the hydrogen ion, but this is the same for all hydrogen halides.
H-Hal(g)..... +.....
water........ ........H+(aq)..... +.....
Hal-(aq)
Once again, we are considering the overall process as the sum of these five processes merely to aid predictions (c.f. section 16.2.5.). This is not the sequence of events. For example, the H-F bond does not break homolytically (section 20.2.1.) producing atoms which then either lose or gain electrons. It breaks heterolytically (section 20.2.2.). But, returning to our artificial division of the processes:
Down group VII:
a) The H-Hal bond becomes weaker which tends to increase acid strength.
b) The halogens show a decreasing ability to gain electrons which tends to decrease acid strength.
c) The halogens show a decreasing attraction for water molecules which tends to decrease acid strength.
In this case the decreasing bond strength outweighs the other two factors, and acid strength increases down the group. Bond strength in this case is more important than was Hal-Hal bond strength in determining the oxidising power of halogens (section 16.2.5.). This must clearly be a function of the hydrogen atom in hydrogen halides.
In fact the small size of the hydrogen atom compared with a second halogen atom makes H-Hal bonds considerably stronger than the corresponding Hal-Hal bonds. It so happens that their greater strength makes the reduction in strength of H-Hal bonds down the group more critical.
These predictions may be checked by using the appropriate enthalpy data in combination with a suitable enthalpy diagram. i.e.
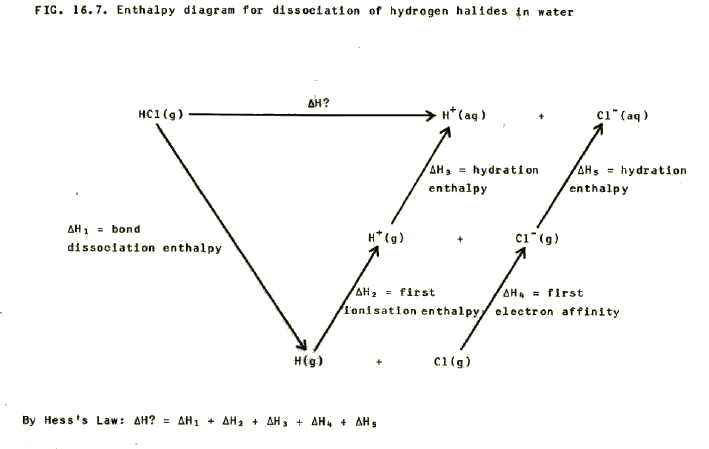
As well as dissolving in water, note that hydrogen halides also dissolve in organic solvents such as methylbenzene, but in this case they dissolve as molcules.
ii) Hydrogen fluoride shows its most complex misbehaviour so far, when it comes to acid strength.
We might predict that hydrogen fluoride will be a particularly weak acid in aqueous solution due to its particularly strong H-Hal bond, and also because fluorine's attraction for an extra electron is less than expected. Indeed, hydrofluoric acid is weak, but there is an additional reason. Hydrogen bonds between one HF molecule and another (section 14.3.1. and FIG. 4.5.) must be broken before protons can be released. The existence and strength of the hydrogen bonds is a characteristic of fluorine's small number of shells combined with that part of its high effective nuclear charge which is due to its high proton:electron ratio.
But hydrogen bonding has a more complex role. Hydrogen fluoride is very soluble due to hydrogen bonding with water molecules. However, the greater the concentration of HF in aqueous solution, the greater the degree of hydrogen bonding between one HF molecule and another. From the argument in the last paragraph this would not be expected to increase dissociation. Indeed, from all we know about weak acids, we would expect dissociation to decrease as concentration is increased. Not so with hydrofluoric acid.
The strength of HF to HF hydrogen bonds takes on an interesting significance as concentration increases. As concentration does increase, hydrogen bonding between HF molecules increases, and so the distinction between hydrogen bond and covalent bond becomes less clear. (c.f. section 16.2.1. covalent vs. van der Waals bonding.) This leads to the formation of [HF2]-, [H2F3]-, [H3F4]-, etc. ions, with simultaneous release of protons.
It is difficult to decide whether these ions are produced via breaks in chains such as those in FIG. 4.5. or via fluoride ions being removed from solution by bonding with HF moleclues etc. The major event presumably depends to some extent on whether water is added to liquid hydrogen fluoride or, for example, the concentration of hydrogen fluoride already in an aqueous solution is increased. However, the fact remains that concentrated solutions of HF are strong acids with high concentrations of readily available protons.
Liquid hydrogen fluoride is also a very strong acid for the same reason. Of course, the mere existence of liquid hydrogen fluoride is a characteristic of its hydrogen bonding.
iii) The redox properties of hydrogen halides have already been discussed in section 16.2.5.vi.
16.3.3. The solubility of some metal halides has already been discussed in section 15.3.3.
The solubility of
silver salts is interesting because of its practical implications (see
appendix i). Silver fluoride is soluble, largely because the strong
electrostatic attraction between water molecules and fluoride ions,
combined with the attraction between water molecules and silver ions,
is sufficient to overcome the electrostatic forces within the lattice.
The change in dissipation of energy (as measured by  S) is
comparatively insignificant.
S) is
comparatively insignificant.
The larger size of the remaining halide ions makes their silver salts insoluble: the weaker attraction of the halide ions for water outweighs the weaker attractive forces in the lattice (cf section 15.3.3.).
TABLE 16.1. The radii (in nm) of some ions relevant to the solubilities discussed here and in section 15.3.3.
| Cations | Anions | ||
| Ion | Radius | Ion | Radius |
| Li+ | 0.068 | F- | 0.133 |
| Na+ | 0.098 | Cl- | 0.181 |
| (Ag+) | (0.126) | Br- | 0.196 |
| K+ | 0.133 | I- | 0.219 |
| Rb+ | 0.148 | ||
| Cs- | 0.167 | ||
Silver chloride, bromide and iodide are so insoluble that they are precipitated from solutions of their soluble salts when aqueous silver nitrate is added. The different colours of the precipitates give a rough indication of which halide is present. Further information is provided by their relative solubilities in dilute ammonia. Silver chloride dissolves readily in dilute ammonia. To redissolve silver bromide, more concentrated ammonia is needed. Silver iodide does not redissolve even in concentrated ammonia solution.
The basis of these differing solubilities can again be traced back to the increasing number of shells in the halide ions as the group is descended. As the halide ions become bigger, their silver salts become even less soluble mainly as a result of the decreasing strength with which the halide ions are hydrated. This means that the concentration of halide and silver ions left in solution after one of the silver halide precipitations becomes even lower as the group is descended.
Furthermore, ammonia redissolves the precipitates by complexing the silver ions and removing them from solution. This pulls more of the salt into solution:
Thus, since complex formation is itself an equilibrium reaction, a sufficent concentration of silver ions must be present for this to occur, and hence the ability of ammonia to redissolve the precipitate becomes less likely as the solubility of the halides decreases.
16.4. MORE ON OXIDATION STATES.
16.4.1. The multiplicity of oxidation states possible with halogens is illustrated clearly by the number of possible halogen-containing anions. Together with the hybridisation of the halogen atom in each case, these anions are summarised in FIG. 16.8...
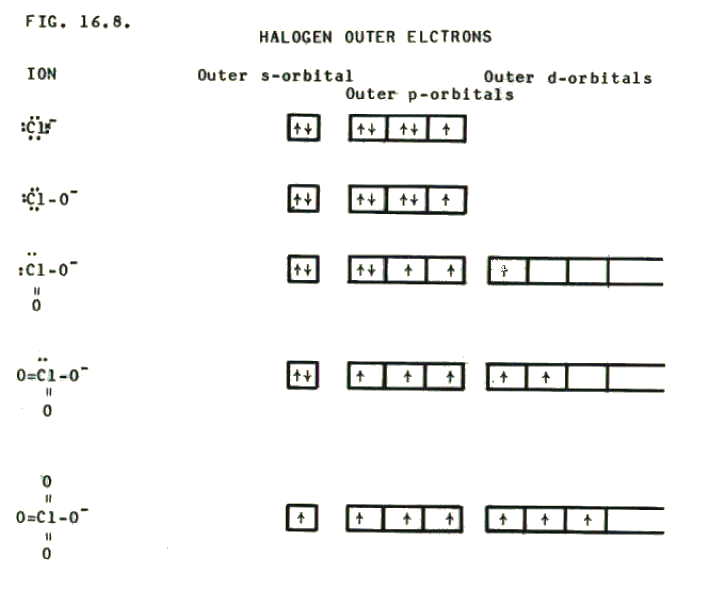
The relative acid strengths of the corresponding acids have already been discussed in section 14.5.3.iv.
16.4.2.i) The relative stabilities of these oxidation states, and the changes in relative stabilities down group VII are demonstrated by the relative stabilities of the above anions in aqueous solution. The stabilities are reflected by the free energies of the anions.
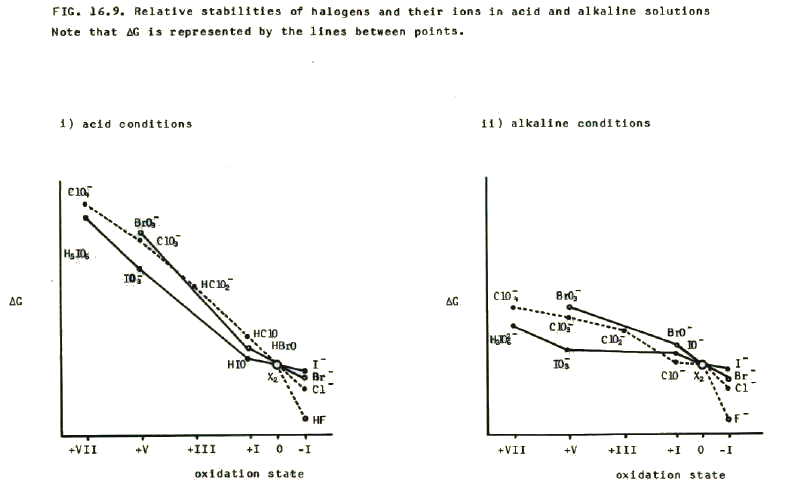
The relative stabilities are in turn demonstrated by the reactions of halogens with water.
There are four possibilities:
a)..... X2(s,l,g)..... +.....
water........ ........X2(aq)
plus:
b)..... X2(aq).. + H2O(l)..... ....H+(aq).. +..
X-(aq)..
+.. HOX(aq)
plus:
c)..... 2HOX(aq)..... .....2H+(aq)..... +.....
2X-(aq)...
+... O2(g)
..................
decomposition
or:
d)..... 3HOX(aq)..... .....3H+(aq)... + 2X-(aq)... +...
XO3-(aq)
...................
disproportionation
There are two main factors that decide which of these reactions occurs:
First, the size (number of shells) of the halogen atom. The further the outer shell from the electrostatic attraction of the nucleus, the more likely it is that the atom can lose control of larger numbers of electrons in covalent bonds i.e. the more likely it is to show higher oxidation states.
Second, the pH of the solution. High concentrations of hydroxide ions remove both weak acids and hydrogen ions from solution, and hence pull the equilibria to the right. This not only encourages the halogens to dissolve by favouring the further reaction of dissolved halogen according to equation b), it also favours reactions c) and d).
Even so, the controlling factors are complex, and predictions are difficult. However, one prediction is simple. Whatever the pH, fluorine is so small that there are no elements more electronegative than it, so it cannot show positive oxidation states. Fluorine does not form oxy-anions and the nett reaction of fluorine with water is b) + c).
i.e....2F2(aq)... + 2H2O(l).... ....4H+(aq)... +...
4F-(aq)...
+....O2(g)
In other words, fluorine oxidises water to oxygen (section 16.2.5.ii.) and is even a strong enough oxidising agent to produce some ozone. (Hydrogen peroxide and oxygen difluoride are by-products.)
The reactions of the other halogens are far more dependant on pH:
ii) In acid or neutral conditions: Reaction c) is favoured over reaction d);
the equilibria are well over to the left, and increasingly so down the group.
Chlorine is a sufficiently powerful oxidising agent to oxidise water, but far more slowly than fluorine oxidises water, presumably because the Cl-Cl is stronger than the F-F bond.
With bromine and iodine, the reaction stops at stage b):
E.g... Br2(aq).. + H2O(l).... ....H+(aq).. +..
Br-(aq)..
+.. HOBr(aq)
Though iodine is hardly even soluble, so even reaction b) is very limited.
iii) in alkaline conditions: reaction d) is favoured over reaction c);
the equilibria are further over to the right than in acid conditions, and increasingly so down the group.
Thus with iodine, the overall reaction is b) + d):
I2(aq).. +..
6-OH(aq).... ....5I-(aq).. +..
IO3-(aq).. +..
3H2O(l)
With bromine, and even more so with chlorine, the reaction tends to stop at stage b):
E.g.. Cl2(aq).. +..
2-OH(aq)... ...Cl-(aq).. +..
-OCl(aq).. +...H2O(l)
unless high concentrations of alkali and/or higher temperatures are used, in which case the nett reaction is b) + d).
iv) Note that formation of halate(VII) ions requires a strong oxidising agent. Only chlorine and iodine are known to form halic(VII) acids. Halate(III) ions are also unstable because it is more favourable for them to disproportionate. Only chlorine is known to form a halic(III) acid.
16.5. QUESTIONS
1) Summarise the changes down group VII of the properties of the halogens and their compounds.
2) How do the changes down group VII compare with the changes down group I. Describe the basis of any similarities or differences between changes down the two groups.
3) Explain which of the halogens will displace iodine from a solution containing iodoide ions. Explain which could be most conveniently used in the laboratory.
4) Predict and draw the shapes of the oxyanions shown in FIG. 16.8. Describe how you predicted these shapes.
5) How is the solubility of silver halides predictable from the ionic radii of halide ions? How does the solubility of silver halides compare with the solubility of group I metal halides?
6) How are the reactions of halogens with i) acid and ii) alkali related to the number of electron shells in the halogen atoms?
Unless
otherwise stated, all materials in this web version of chapter 16 are ©
2007 Adrian Faiers MA (Oxon) MCIPR

What 's the connection between a dozen eggs and
a garden mole?

Answer: Not a lot, really, but see Chapter 1