Recommended
by:
Top
20 UK science resources,
The Tutor Website
Recommended by:

Rated:

2010
Listed on the science,
engineering
and technology section of 
'providing you with access
to the very best Web resources for education and research, evaluated
and selected by a network of subject specialists.'
(Please note that intute closed in July 2011)
Section 3: Organic
CHAPTER
22: PREDICTING REACTIONS [ II ]
(Oxygen and nitrogen
containing compounds)
NB This chapter has now been
updated to improve browser compatibility.
Please
use the 'send email' link at the top right hand corner of this page to
report any problems.
QUICK SKIPS:
(Click on the 'back' arrow to get back to this quick skip section)
Alcohols
Ethers
Carbonyl compounds
(Aldehydes and ketones)
Carboxylic
acids and their derivatives
Amines
Diazonium Compounds
Nitriles
Nitro-compounds
22.1.1. Predictions i) Oxygen is more electronegative than carbon, and the C-O bond in alcohols is polarised, exposing nuclear charge on the functional carbon atom. This makes the carbon susceptible to nucleophilic attack. -OH is not a very good leaving group, but attack nevertheless results in substitution of the OH group. The overall reaction is therefore nucleophilic substitution via mechanisms 2 and 3 (FIG. 20.1.).
22.1.2. Some general points about leaving groups. We saw in the last chapter that the halogens leave with decreasing ease in the order I > Br > Cl > F. The reason is this. During the process of leaving, the outer electron cloud of the halogen becomes distorted. Distortion is easier, when the halogen is large and the outer shell is far from the influence of the nucleus. Moreover, the larger the resultant anion, the less concentrated is the negative charge.
More generally, uncharged or neutral moleclues are better leaving groups than ions. Thus ease of leaving decreases in the order H2O >> -OH > -OR.
In fact, the hydroxide ion is such a poor leaving group that it can not be simply replaced many nucleophiles such as halide ions.
However, protonation of the OH group changes the leaving group to water, an excellent leaving group. Simple nucleophilic substitution of alcohols must therefore be performed in acid conditions.
22.1.3. i) The most commonly encountered nucleophiles to react with alcohols are therefore halide ions in acid conditions. The reagent used most commonly is the halide plus sulphuric acid, rather than the hydrohalic acid itself.
However, other reagents are often used to substitute halogen atoms in place of the OH group. Phosphorus trihalides, phosphorus pentahalides and sulphur dichloride oxide are the most commonly encountered halogenating agents.
Often, phosphorus trihalides are created in situ by using iodine/bromine and red phosphorus, and the conversion of alcohols into iodoalkanes by red phosphorus and iodine is a common starting reaction in synthetic pathways. In such pathways the iodoalkane is then frequently used for reaction with other nucleophiles. This is because the two steps together usually give a better yield than treating the alcohol directly.
Reactions with these halogenating agents still occur via a nucleophilc mechanism, though the nucleophile is not a simple halide ion. Equations for the reactions are as follows:
3ROH.... +....
PHal3.......![]() .......
3RHal.... +.... P(OH)3
.......
3RHal.... +.... P(OH)3
ROH...
+... PHal5.......![]() .......
RHal... +... HHal...
+...
POHal3
.......
RHal... +... HHal...
+...
POHal3
ROH....
+.... SOCl2......![]() ......
RCl... +... HCl...
+...
SO2
......
RCl... +... HCl...
+...
SO2
The HCl gas evolved in the reaction with phosphorus pentachloride dissolves in the moisture present in air to produce steamy white fumes of hydrochloric acid. This is often regarded as a test for the presence of the alcohol groups.
ii) Another nucleophile to react with protonated alcohols is a second alcohol molecule. The conditions for this are seen in fig 22.1 below.
22.1.4. Effects of the rest of the molecule. Apart from the considerations in section 22.1.5. below, an important effect on nucleophilic substitution of the OH groups occurs when they are attached to benzene rings. As with halobenzenes, phenols are resistant to nucleophilc substitution i) because the high electron density of the ring prevents approach by nucleophiles, and ii) because delocalisation of a lone pair from the hydroxyl oxygen into the ring increases the electron density around the carbon atom and also gives the C-O bond partial double character.
22.1.5. Predictions ii) As with haloalkanes, there is another possibility which may accompany the loss of the leaving group. A hydrogen may be lost from the carbon atom next to the functional carbon. The overall reaction is therfore elimination via mechanisms 10 and 11 (FIG. 20.1.).
However, it is not
possible to use a strong
base in this case, because acid conditions are needed to aid loss of
the OH group. The reagent is concentrated sulphuric acid. There are
several possibilities for reaction of alcohols with sulphuric acid and
they are summarised for ethanol in the scheme below.
(Click on diagram to
enlarge
on a new web page)
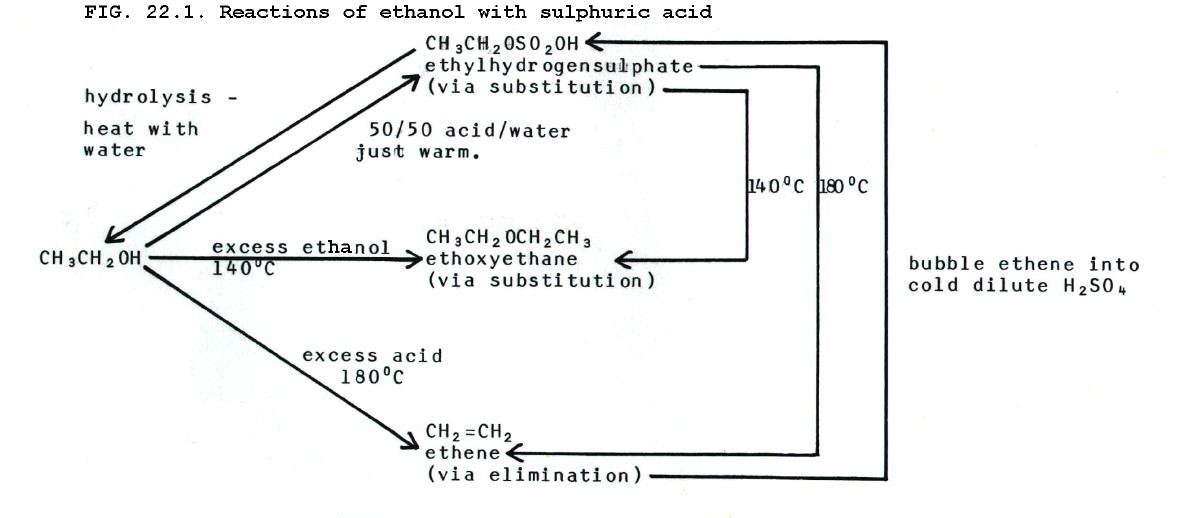
{Note that the alcohol acts both as the attacked species and
the attacking reactant in the etherification. Excess alcohol is
necessary so that some is protonated and attacked and some is not
protonated (attacking).}
Thus, with sulphuric acid, alcohols show the same competition between substitution and elimination as that shown by haloalkanes with potassium hydroxide. In this case the factors which favour elimination are:
i) Branching around the functional carbon atom.
ii) Excess acid.
iii) Higher temperatures.
Compare this with the
list
for haloalkanes (section 21.7.4). There is also the same competition
between unimolecular and bimolecular mechanisms for substitution, but
note the mechanism for acid catalysed elimination is:
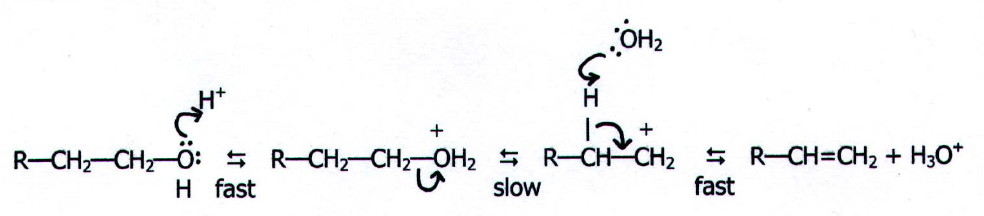
The concentrated sulphuric acid not only acts as a catalyst,
but also acts as a dehydrating agent in the second step.
22.1.6. Predictions iii). The available lone pairs on the hydroxyl oxygen enable alcohols to act as bases. The same property of alcohols enables them to act as nucleophiles. Alcohols are therefore able to act as bases or nucleophiles.
The positive inductive effect of the alkyl group makes alcohols slightly stronger bases than water. However, nucleophilic strength is also affected by steric factors. Thus an alcohol with a lot of branching around the hydroxyl group might not be a very effective nucleophile, because the carbon chains physically (sterically) hinder it from reaching the exposed nuclear charge.
22.1.7. The basic properties of alcohols are best demonstrated by i) the importance of protonation in nucleophilic of the OH group, and
ii)
the hydrogen
bonding
which makes simple alcohols liquids at room temperature.
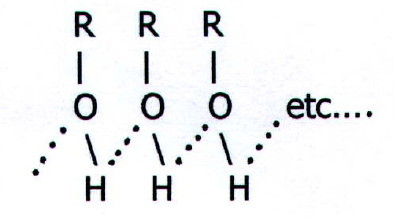
Note that hydrogen bonding between water and the OH group of simple alcohols also accounts for the solubility of simple alcohols in water. When hydrogen bonding with organic molecules is not possible, solubility in water is unlikely. Even as the non-polar part of a molecule containing a group which can hydrogen bond becomes larger, so solubility decreases.
22.1.8. i) The best known example of alcohols acting as nucleophiles is esterification (section 22.3.6.ii.).
ii) Esterification is not to be confused with etherification, another important example of alcohols acting as nucleophiles. The conditions for etherification, using the alcohol as the only organic reagent, are shown in fig 22.1.
However, alkoxide and phenoxide anions are far better nucleophiles than the parent alcohols. The ions are produced by dissolving sodium in the alcohol/phenol (section 22.1.10.ii.). A second organic reagent is used, a haloalkane, and it is this which undergoes nucleophilic substitution.
RO-..... +.....
R'Hal.........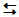 .........
R'-O-R..... +..... Hal-
.........
R'-O-R..... +..... Hal-
The molecularity of the reaction is determined largely by the nature of the alkyl group in the haloalkane.
22.1.9. Predictions iv). The electronegativity of the oxygen atom enables it to carry negative charge and alcohols are thus able to lose the hydroxyl hydrogen atom. They are therfore able to act as acids, making alcohols amphoteric, like water.
22.1.10. Examples of acidic behaviour in alcohols:
i) Reaction with hydroxides. A solution of potassium hydroxide in ethanol (sodium hydroxide is insoluble) contains ethoxide ions:
-OH..... +.....
EtOH..................
H2O.....
+..... EtO-
ii) With alkali metals E.g.
2EtOH.....
+..... 2Na.........![]() .........
2NaEtO..... +..... H2
.........
2NaEtO..... +..... H2
22.1.11. Effects
of
the rest of the molecule on acidic behaviour of the OH group.
The acidic strength of phenols is even greater than that of aliphatic
alcohols. This is because the negative charge in the resultant
phenoxide ion is spread over the benzene ring by delocalisation:
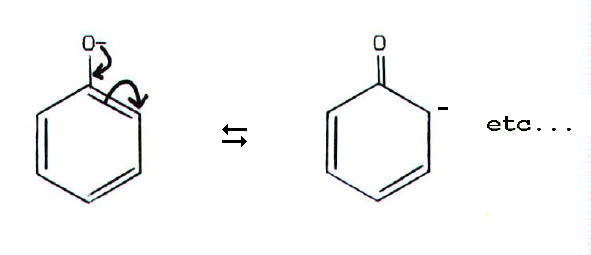
(Note that delocalisation
of a lone pair into the benzene ring, makes phenols weaker bases than
aliphatic alcohols.)
Thus phenols not only react like aliphatic alcohols with alkali and alkali metals, but also they dissolve in water to produce acidic solutions (pKa for phenol = 9.98). However, this pKa value shows that they are not strong enough acids to release carbon dioxide from carbonates.
Note also that the benzene ring in phenols is activated and susceptible to electrophilic substitution in the 2,4 and 6 positions.
22.1.12. Other reactions.
i) Oxidation. Alcohols are sometimes used as fuels
2C2H5OH..... +.....
4O2.........![]() .........
2CO2.....
+..... 6H2O
.........
2CO2.....
+..... 6H2O
They can also be
oxidised by
a variety of means to aldehydes, ketones, and/or carboxylic acids,
depending on the type of alcohol and the means of oxidation. The
possibilities are summarised in Fig 22.2. below.
(Click on diagram to
enlarge on a
new web page)
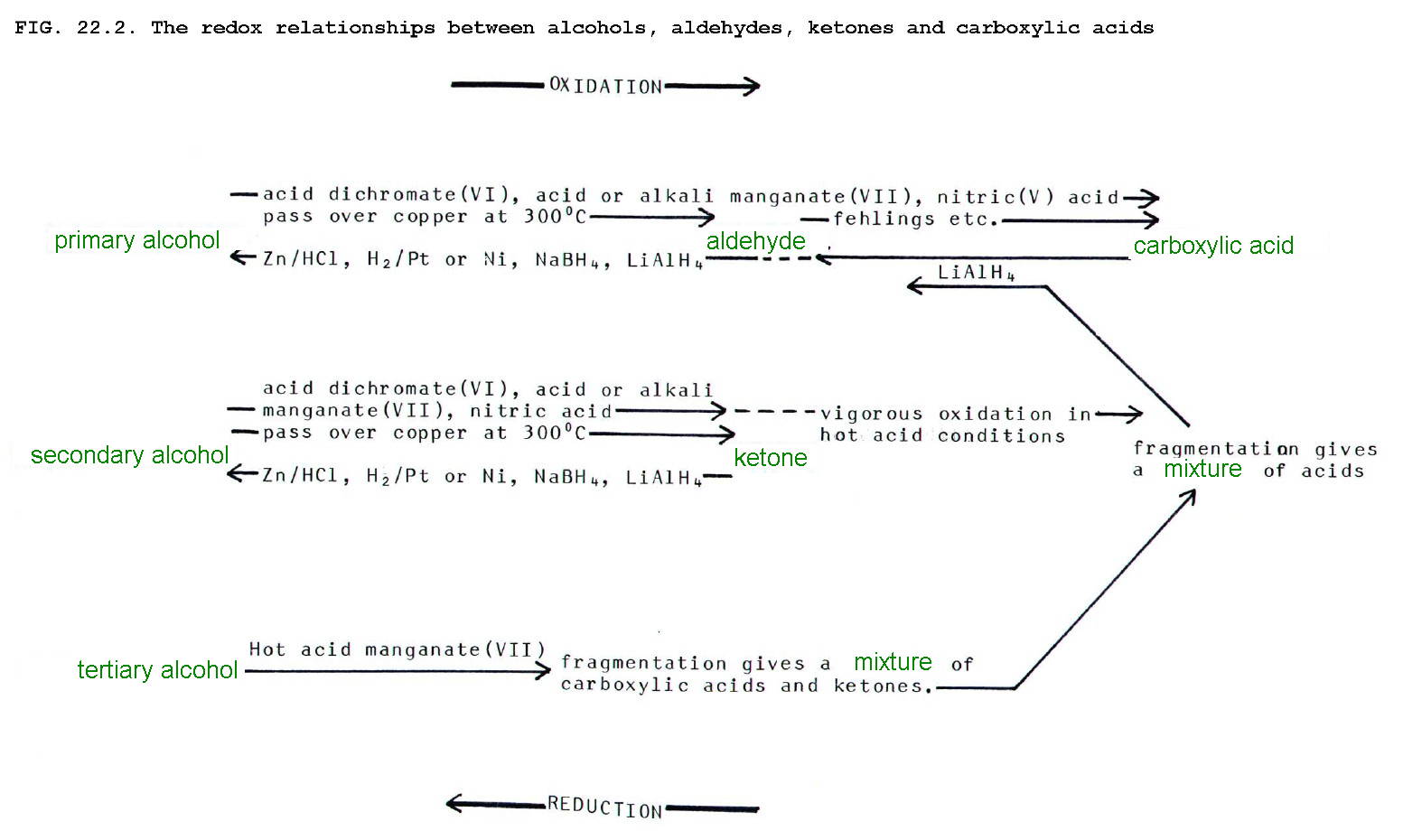
It is difficult to stop the oxidation of primary
alcohols at the aldehyde stage and dehydrogenation over heated copper
is thus a particularly useful reaction since the aldehyde does not
react further. The reaction is used industrially.
It is possible to prepare aldehydes using the more conventional laboratory reagents simply by choosing an appropriate technique. To obtain the aldehyde, it must be removed from the reaction mixture before it can be further oxidised.
Thus
alcohol is dripped into boiling acidified dichromate(VII) and the
aldehyde distilled off as it forms. In contrast refluxing takes the
reaction to the carboxylic acid stage:
(Click on diagram to
enlarge on a
new web page)
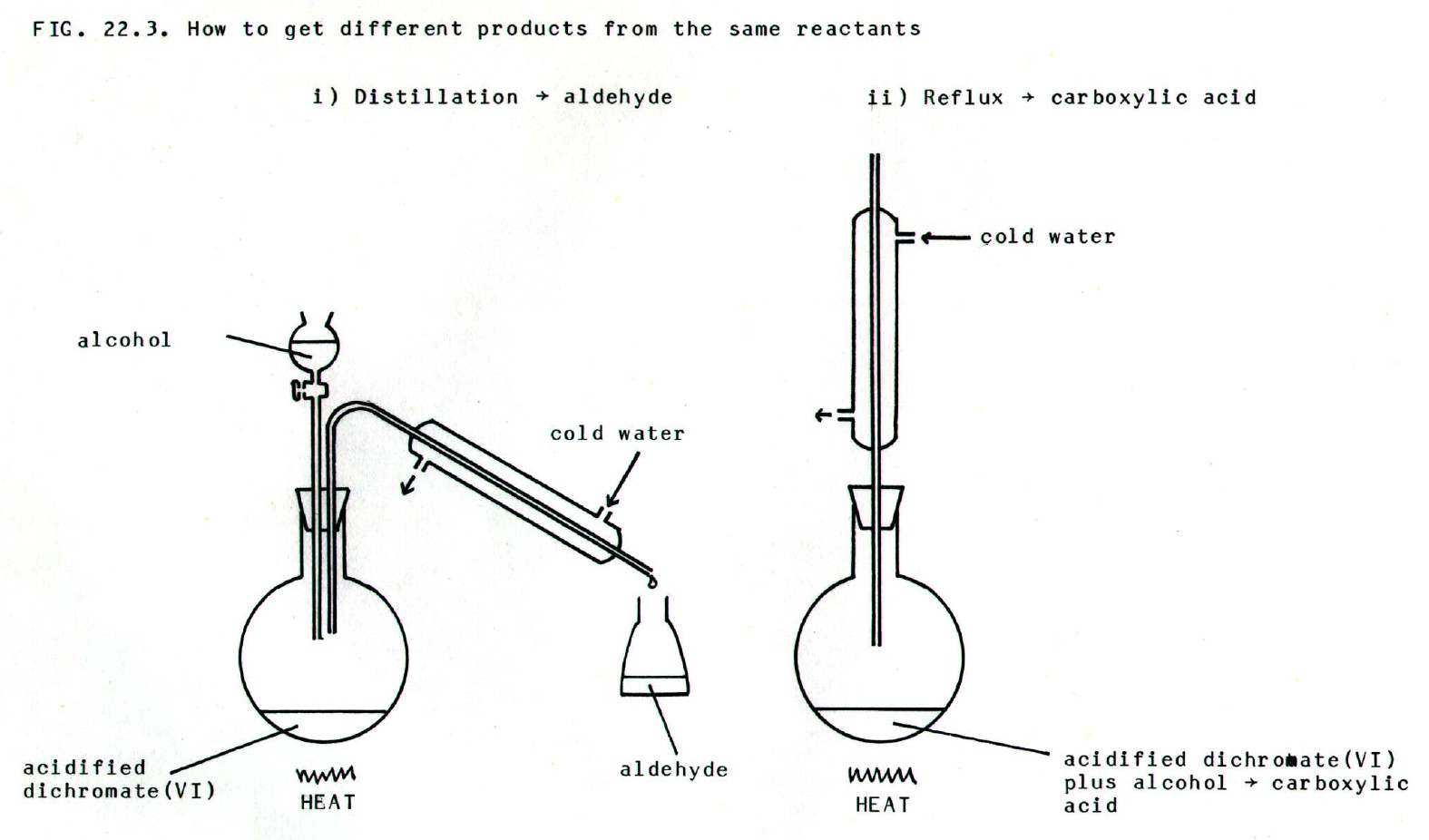
ii) The Haloform reaction.
Under the reaction conditions used for the haloform reaction, primary
and secondary alcohols are oxidised respectively to aldehydes and
ketones(section 22.2.6.iv.).
Thus, both secondary alcohols with at least one methyl group attached to the functional carbon, and ethanol give the haloform reaction.
iii) Iron(III) chloride: In neutral solution, phenols produce a purple colouration with iron(III) chloride (normally orange). The new colour is due to replacement of the iron(III) ligands.
Ethers are less soluble
in
water than are the corresponding alcohols, but they are more soluble in
acids. This can be understood because ethers have an extra alkyl group
exerting a positive inductive effect. This makes the lone pair on the
functional oxygen more available.

Aromatic ethers are not such strong bases because the lone
pairs delocalise into the benzene ring (compare with phenols 22.1.11.)
Ethers are generally much less reactive than alcohols. They have no available hydrogen and do not react with sodium in the cold, nor do they react with phosphorus pentachloride in the cold. They do, however, undergo nucleophilic substitution with concentrated hydriodic acid.
R-O-R.....
+..... HI.........![]() .........
2RI..... +..... H2O
.........
2RI..... +..... H2O
22.2. CARBONYLS
(ALDEYHDES AND KETONES)
22.2.1. Predictions i). Oxygen is electronegative, meaning that the electrons in the C=O double bond are concentrated more on the oxygen. Thus the functional carbon's nuclear charge is exposed to attack by nucleophiles.
Moreover, the C=O bond is unsaturated and attack results in addition. The overall reaction is therefore nucleophilic addition via mechanism 7 (FIG. 20.1.).
22.2.2. Nucleophiles which add to carbonyls
| Table 22.1. Nucleophiles which add to carbonyls (Al indicates aldehyde product, Ke indicates ketone product) | |||
| Nucleophile | Reagent | Conditions | Main organic products |
| AlH4- or BH4- | LiAlH4 or NaBH4 | Warm gently in DRY ether | Al Ke |
| R- | Grignard reagent: RMgX | In DRY ether. Warm to start reaction. | Methanal Other Als Ke |
| d+.. d- R - OH |
Alcohol | In dilute acid (e.g. dil. HCl) | Al Ke do not readily react |
| SO3- | Sodium hydrogen- sulphite | cold | Al&Ke crystalline bisulphite compounds. Readily isolated and carbonyl restored by acid or alkaline hydrolysis, thus reacn. used to purify carbonyls. |
| -CN | NaCN
+
dil. H2SO4 (g HCN) |
cold | Al&Ke |
| NH3 | Ammonia | In ether O°C | Al Ke no reaction |
Notes:
Note i) Examples of:
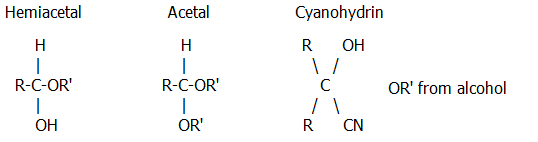
Note ii) pH is often critical since reaction is aided by protonation of the carbonyl, and yet free nucleophile must also be present. This is an example of a use for buffers.
22.2.3. Effect
of
the rest of the molecule. The rate of
nucleophilic attack on the carbonyl carbon depends on the extent to
which nuclear charge is exposed. Thus alkyl groups reduce the positive
charge owing to their electron pushing (+I) effect. It follows that
reactivity decreases in the order:
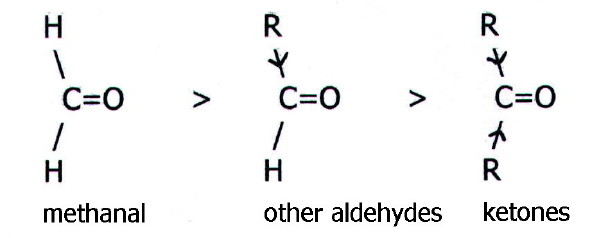
Large alkyl groups particularly hinder the approach of
nucleophiles.
Moreover,
if the functional carbon is next to a double bond or benzene ring, the
concentration of positive charge is reduced by overlap of the p-bonding systems:
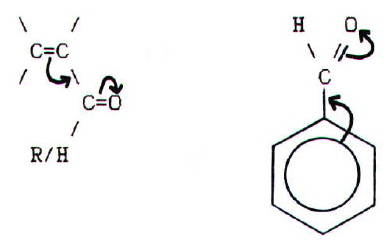
In addition, the rest of the molecule may have an even more
profound effect on a carbonyl's reactions. (E.g. See the aldol
condensation, haloform reaction, and Cannizzaro's reaction below.)
22.2.4. Predictions ii) Addition may result in two electron withdrawing groups being attached to the same carbon atom. Often when this occurs, the addition moleclue breaks down eliminating a small molecule such as water. The overall reaction is therefore addition-elimination and it is particularly common when the adding molecule has the general formula :NH2X.
22.2.5. Addition-elimination in carbonyls: some examples.
| Table 22.2. Some nucleophiles which give addition-elimination reactions with carbonyl compounds. | ||
| Nucleophile | Conditions | Main organic product |
| Hydroxylamine (:NH2-OH) |
warm | oximes (aldoximes and ketoximes) |
| Hydrazine (:NH2-NH2) |
room temp. or warm | hydrazones |
| Phenylhydrazine (:NH2-NHC6H5) |
room temp. or warm | phenylhydrazones |
| *2,4-dinitro- phenylhaydrazine |
room temp. | *2,4-dinitrophenylhydrazones
= yellow crystalline solids. Reaction used as test for presence of carbonyl group |
| Semicarbazide (:NH2.NHCO.NH2) |
room temp. | semicarbazones |
| Amines | room temp. | "Schiff's" bases |
| Carbonyl
compounds with at least one H on the C next to the carbonyl C |
alkali or
stronger base, gentle warmth |
†see below |
Notes:
Note i) * These 2,4-dinitrophenylhydrazones (and 4-nitrophenylhydrazones) are easily purified by crystallisation techniques. They also have characteristic and sharp melting points. Thus the reactions with nitrophenylhydrazines are used in combination with melting point determination to identify carbonyl compounds.
Note ii)
† a) The products
of this reaction vary according to the reacting carbonyls. The first
step is removal of the a-hydrogen
by the base:
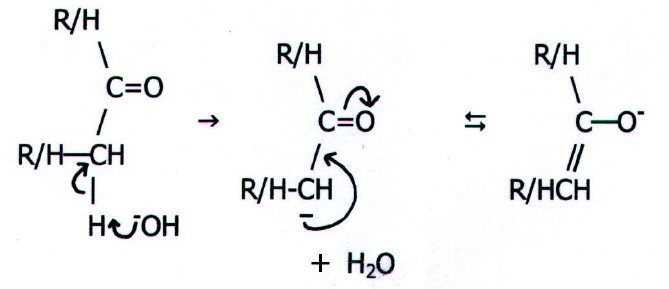
It can
be seen that delocalisation of the negative charge over the carbonyl
group and its electronegative oxygen atom, is what makes this possible.
The carbanion now acts as a nucleophile:
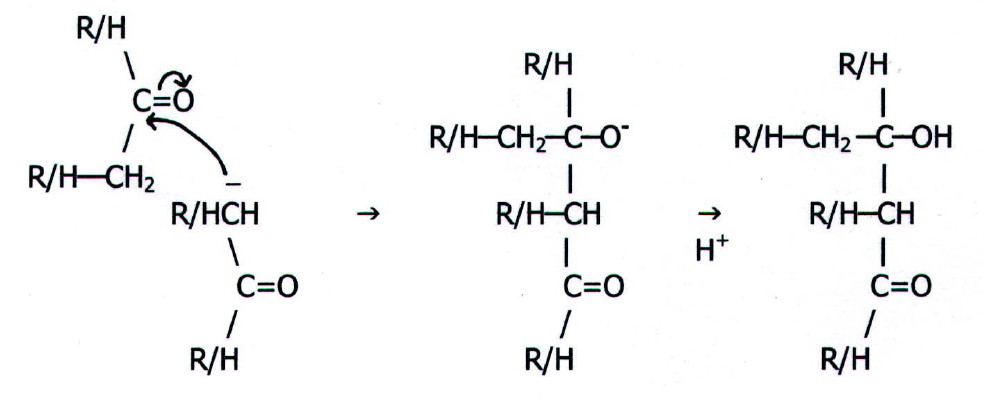
These large addition products readily undergo elimination:
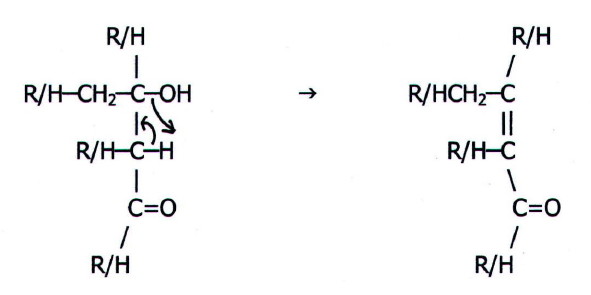
The products are
themselves carbonyl compounds and can undergo further addition. In the
case of propanone and short chain aliphatic aldehydes, continued
addition can be extensive, producing polymers of high relative
molecular mass. These are observed as brown resins.
b) When there is
no 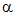 -hydrogen, there is still a
reaction in alkaline conditions. First, the molecule can still undergo
the above reaction as long as there is another carbonyl present which does
have an -hydrogen.
-hydrogen, there is still a
reaction in alkaline conditions. First, the molecule can still undergo
the above reaction as long as there is another carbonyl present which does
have an -hydrogen.
If there is not another
such
carbonyl present, aldehydes (not ketones) with
no -hydrogen undergo
disproportionation. Eg.
(This
is often referred to as Cannizzaro's reaction.)
22.2.6. Other reactions
i) Oxidation
Like most organic compounds, both aldehydes and ketones burn in air to produce carbon dioxide and water. However, the similarity between the oxidation reactions of aldehydes and those of ketones ends here.
In general, aldehydes are much easier to oxidise than ketones, as summarised in figure 22.2 above. They are, then, readily oxidised to the corresponding carboxylic acids by acidified dichromate.
............................
acidified dichromate(VI)
RCHO.....
+..... [O]................![]() ................
RCOOH
................
RCOOH
Not only do aldehydes react with acidified dichromate, they are also oxidised by ammoniacal silver nitrate (Tollen's reagent), Fehling's solution (containing complexed copper(II) ions in alkaline solution), and Schiff's reagent (rosaniline dye decolourised by treatment with sulphur dioxide). Like the reaction in the above equation, and like the reaction with the more vigorous oxidising agent, manganate(VII), there are distinctive observable changes:
| Table 22.3. Observable changes when aldehydes react with various oxidising agents | |
| Description
of oxidising agent (plus brief conditions) |
Description of reduction product |
| Acidified
dichromate: orange solution (heat) |
Complexed
chromium(III) ions: green to blue soln., depending on ligands |
| Acidified
manganate(VII): deep purple solution (warm) |
Complexed
magnesium(II): colourless solution |
| Alkaline
manganate(VII): deep purple solution (warm) |
Manganese(IV) oxide: brown precipitate |
| Diamminesilver
ions: colourless Tollens solution (water bath) |
Silver:
forms as metal mirror on sides of CLEAN test tube |
| Complexed
copper(II) ions: deep blue Fehling's solution (heat) |
Copper(I) oxide: red/brown precipitate |
| Schiff's
reagent: colourless solution (warm) |
Magenta (deep pink) solution |
In contrast, ketones are oxidised only by heating with the strongest of the above oxidising agents, acidified potassium manganate (VII). Under these vigorous conditions, the carbon chain is fragmented to give a mixture of carboxylic acids.
ii) Reduction
FIG. 22.2. also summarises the reduction reactions of carbonyls. Both aldehydes and ketones are reduced to the corresponding alcohol by a variety of reducing agents. These include lithium tetrahydridoaluminate(III) or sodium tetrahydridoborate(III) in dry ether (see nucleophiles in TABLE 22.4.), hydrogen over a hot platinum or nickel catalyst, sodium in ethanol, zinc in hydrochloric acid, and sodium amalgam in water.
Aldehydes give the corresponding primary alcohol, and ketones give the corresponding secondary alcohol:
...................................
Pt
RCHO..... +..... H2.........![]() .........
RCH2OH
.........
RCH2OH
..................................
heat
..
R...............................................
R
.... \ ..................................
Pt......... \
..... C=O..... +.....
H2...........![]() ...........
CHOH
...........
CHOH
.... /.................................
heat........ /
.. R...............................................
R
Stronger reducing agents, such as hydrazine followed by a strong base, or zinc amalgam in concentrated hydrochloric acid, reduce carbonyls further to hydrocarbons:
....
\..............................
\
..... C=O...........![]() ...........
CH2
...........
CH2
.... /.............................. /
This is useful in conjunction with acylation for introducing a single hydrocarbon chain into a benzene ring (TABLE 21.3.).
iii) Other redox reactions
Disproportionation by certain aldehydes in alkaline conditions has already been discussed in the notes following TABLE 22.2.
iv) The haloform reaction
We have already seen that observable differences in various reactions of carbonyls (such as reactions in alkali and oxidation reactions) allow them to be used as distinguishing tests. Some of the tests allow one carbonyl to be distinguished from another, some provide evidence for the presence of a carbonyl as opposed to another type of compound.
The haloform reaction, and particularly the iodoform reaction, is another useful distinguishing reaction. It is a reaction of methyl groups attached to carbonyl carbon atoms.
However, under the reaction conditions, alcohols are oxidised to carbonyl compounds so all the following compounds give the haloform reaction:
| Table 22.4 Organic compounds which give the haloform reaction | |
| alcohols | carbonyls |
| ethanol
(normal
starting material in preparation of trichloromethane) |
ethanal |
| secondary
alcohols with a methyl group on the funtional carbon i.e...... CH3 ........... \ ............ CHOH .......... / ..... R/H |
methyl
ketones ......... CH3 .......... \ ........... C=O .......... / ..... R/H |
The inorganic reactant in the haloform reaction is XO-, where X is chlorine, bromine, or iodine. Also, hydroxide ions are needed for the final hydrolysis step.
The reagent can be:
a) an aqueous solution
of
sodium oxohalate (I) plus a little alkali
b) the halogen
dissolved in alkali
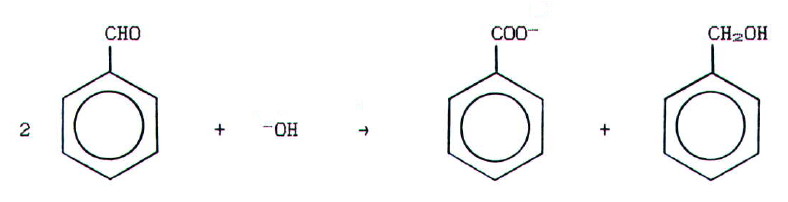
The reaction occurs in two parts:
substitution of methyl hydrogens
...
CH3...............................................
CX3
....
\....................................................
\
..... C=O..... +.....
3-OX...........![]() ...........
C=O..... +..... 3-OH
...........
C=O..... +..... 3-OH
.... / ...................................................
/
R/H................................................
R/H
followed
by hydrolysis
Chloroform
and bromoform are colourless oily liquids and iodoform is a pale yellow
crystalline solid. All the haloforms are alleged to have a "sweet"
smell.
{A
key factor in the reaction is that the intermediate in the first step
is stabilised by delocalisation:

Note that this represents acidic behaviour by a-hydrogen
atoms in carbonyl
compounds - not the only example (TABLE 22.2. note ii.).}
v) Halogenation
This is yet another reaction with useful distinguishing features. Carbonyl compounds react with halogenating agents like phosphorus pentachloride, phosphorus trichloride etc. to give gem-dihalides (two halogen atoms on the same carbon atom):
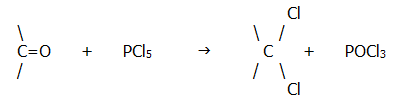
However, in the reaction with PCl5 no HCl is produced, so the reaction can be distinguished easily from the reaction with alcohols (section 22.1.3.).
vi) Polymerisation reactions
Apart from the polymerisation reaction described in the notes following table 22.2., aldehydes in particular undergo various other polymerisation reactions.
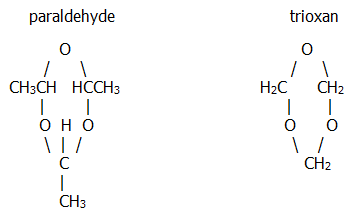
a) In concentrated sulphuric acid at room temperature ethanal gives a liquid trimer, "paraldehyde", used as a hypnotic. At O°C it gives a tetramer, "metaldehyde", used as a slug bait and as a solid fuel for picnic stoves.
Methanal forms the crystalline trimer, "trioxan" on boiling with dilute sulphuric acid:
b) With limewater, methanal first gives the dimer, hydroxyethanal, and then gives a mixture of monosaccharide sugars called "formose". It would be nice to think this is an important biochemical reaction, for example in photosynthesis, but it is not.
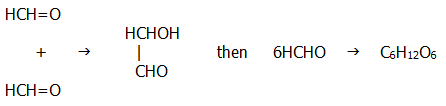
22.3. CARBOXYLIC
ACIDS AND
THEIR DERIVATIVES
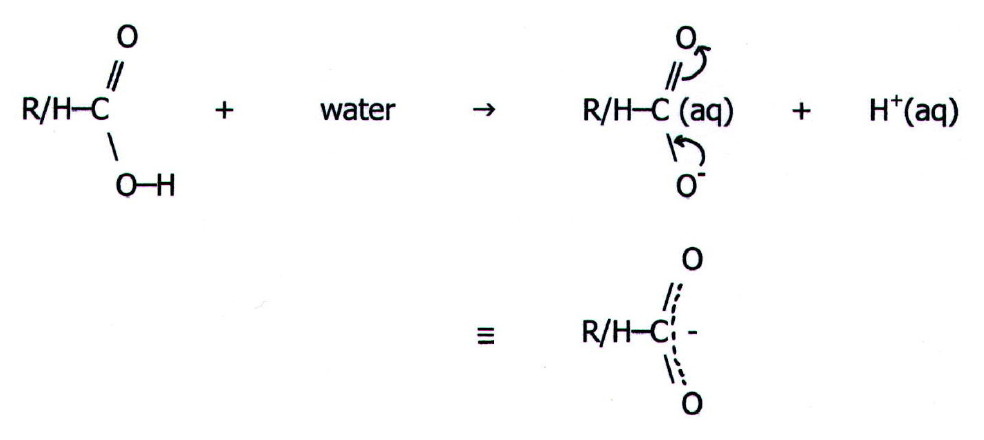
22.3.1. Predictions i) Following the emphasis on twelve main reaction types (chapter 20) it would be tempting to start off by predicting that carboxylic acids and their derivatives undergo two-step bimolecular nucleophilic substitution reactions. Indeed they do. However, as the name implies, carboxylic acids are most noticeably acids.
This is first because
the electronegative
oxygen atoms pull electrons towards themselves. This weakens the O-H
bond, allowing it to break more easily. Secondly, the negative charge
in the resultant anion is delocalised over the two electronegative
oxygens in the carboxylate group. This is a thermodynamic effect which
allows us to predict that the equilibrium will be further in the
direction of anion formation than would otherwise be expected:
This makes the OH group in
carboxylic acids not only more acidic than that in alcohols, but also
more acidic than the OH group in phenols. Carboxylic acids are
therefore weak acids in absolute terms (section 10.2.2.) though strong
compared with other organic hydroxy-containing compounds.
22.3.2. Examples of acidic behaviour
i) Carboxylic acids dissociate in water, as shown in the above equation, producing aqueous hydrogen ions.
ii) With the reactive metals, aqueous solutions of carboxylic acids release hydrogen:
Eg. 2RCOOH(aq).
+..2Na(m)... ...
2RCOO-(aq).
+.
2Na+(aq).
+..H2(g)
iii) Unlike alcohols and phenols, carboxylic acids are stronger acids than carbonic acid and they release carbon dioxide from carbonates and hydrogen carbonates:
Eg. 2RCOOH(aq).
+..2CO32-(aq). .
2RCOO-(aq)..+..CO2(g).
+.
H2O(l)
The reaction with aqueous sodium carbonate is taken as evidence for the presence of carboxylic acids. Like any evidence, it is most useful in combination with other evidence.
iv) Carboxylic acids can be released from their salts by strong mineral acids. This is as much to do with relative volatilities and solubilities as it is to do with relative strengths of acids:
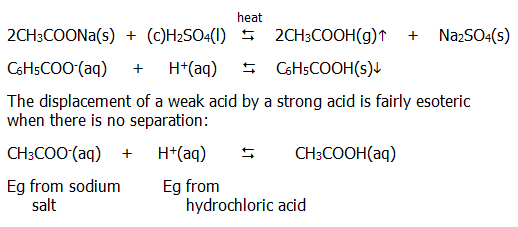
22.3.3. Effects of the rest of the molecule on acid strength.
From the prediction of acidic behaviour in section 22.3.1., it is easily possible to go on and predict how acidic strength is affected by the rest of the molecule.
i) In general, electron pushing groups attached to the carboxyl carbon reduce acid strength by reducing the ease with which the critical O-H bond breaks and by increasing electron density in the resultant anion.
Thus acid strength
decreases
in the series:
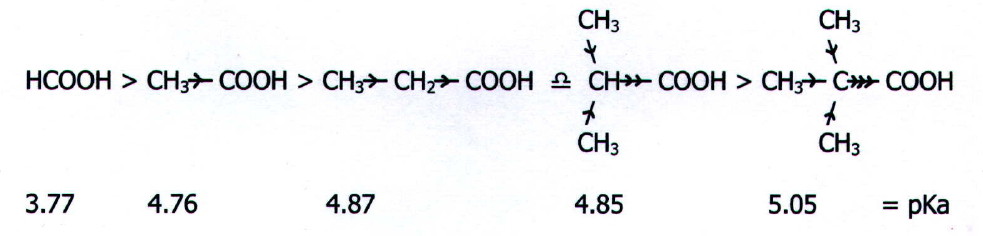
Note
also that the electron pushing effect of a benzene ring one removed
from the carboxyl carbon atom gives phenylethanoic acid a pKa value of
4.31.
ii) Conversely, electron withdrawing groups increase acid strength by increasing the ease with which the O-H bond breaks and by decreasing electron density in the resultant anion.
Thus acid strength
decreases
in the series:
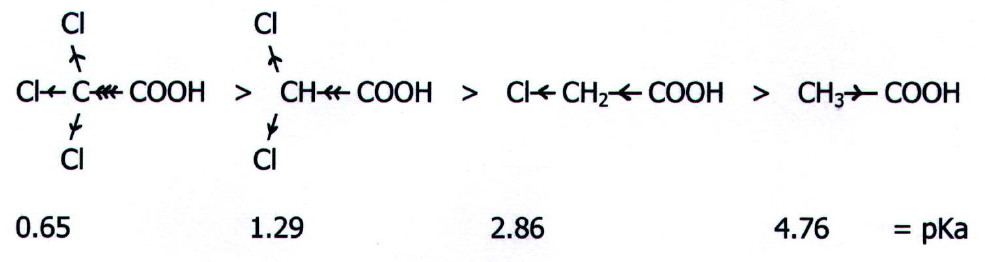
For
comparison, the pKa value for fluoroethanoic acid is 2.66, for
bromoethanoic acid it is 2.90, and for iodoethanoic acid it is 3.17.
Apart from these
inductive
effects, Acid strength may also be increased by negative mesomeric
effects. Thus benzoic acid has a pKa of 4.20, compared with a pKa for
cyclohexylmethanoic acid of 4.87:
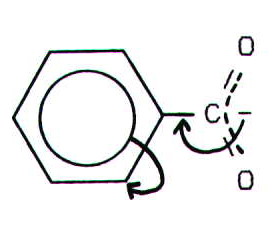
However, use of the Kekulé model suggests that electrons are delocalised OUT of the benzene ring rather than INTO the benzene ring:
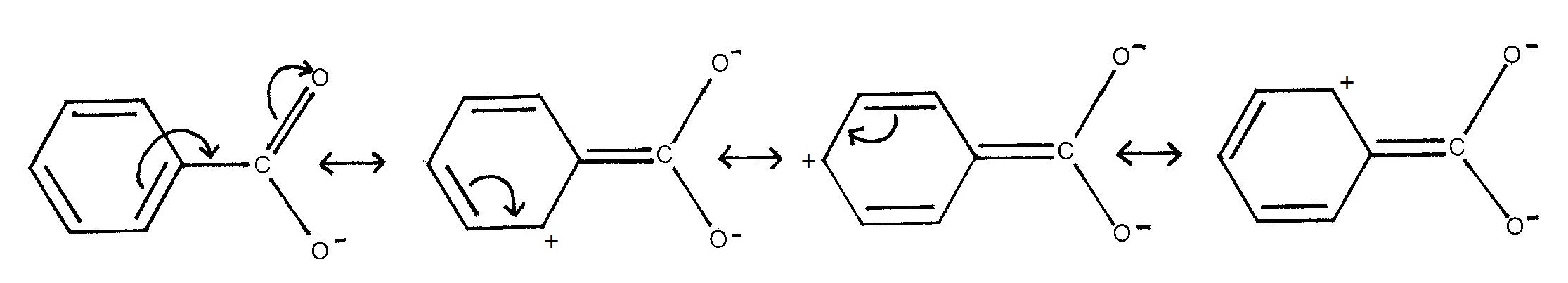
It could be argued that there is still delocalisation over the C-C bond, but the generally accepted argument (which is beyond the scope of study at this level) is that the greater the s contribution to the hybridisation of the adjacent carbon (alpha carbon) the more closely the electrons are drawn to the nucleus of that carbon atom. Arguments based on this model have the advantage that they can also be used to make predictions about the acid strength of carboxylic acid groups bound to doubly bonded and triply bonded carbons, since sp1 carbons are predicted to be considerably less electron donating than sp2 carbons which are, in turn less electron donating than sp3 carbons.
22.3.4. Predictions ii) The functional carbon atom in carboxylic acids and their derivatives has two electronegative groups attached to it. This exposes nuclear charge on the functional carbon atom and makes it susceptible to nucleophilic attack.
Since the carboxyl
group contains a C=O double
bond, it might be expected to undergo addition as in carbonyl
compounds. However, such addition would involve destruction of the
following delocalisation, and would therefore involve concentration of
electron density:
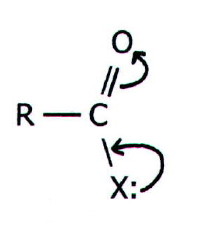
There
is another factor which supresses addition in carboxylic acids,
themselves. In the liquid phase and in solution, they exist either as
ions in which negative charge is delocalised over the whole carboxylate
group, or they exist as dimers which would need breaking before
addition could occur:
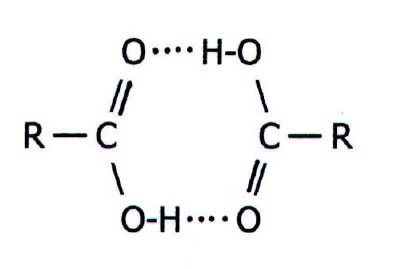
Obviously, this second factor affects any reaction
of carboxylic acids, but the reaction which does
follow nucleophilic attack is substitution of the X
group. This occurs only with nucleophiles which maintain the
carboxylate delocalisation. i.e. there is a lone pair on the new X
group. The only exception is reduction by LiAlH4
or NaBH4. In this case the particularly strong
bond with the small substituting hydrogen atom, and the powerful
reducing qualities of the reducing agent, compensate for the loss of
delocalisation.
The overall reaction is two-step bimolecular nucleophilic substitution via mechanism 4 (FIG. 20.1.).
22.3.5. Relative reactivities of carboxylic acids and their derivatives.
The carboxylate anion is not susceptible to nucleophilic attack because negative charge is delocalised all over the group. Thus, carboxylic acids do not undergo nucleophilic substitution in alkaline conditions.
Even when associated, carboxylic acids are not particularly reactive because the OH group is not a very good leaving group. However, nucleophilic substitution is acid catalysed because protonation of the OH group converts it into the far better leaving group, H2O. Protonation also enhances the leaving ability of the OR group in esters and the NH2 group in amides.
The two main factors which enable prediction of the relative reactivities of carboxylic acids and their derivatives are:
i) The relative
abilities of different X
groups to act as leaving groups
ii) The relative exposure of
nuclear charge on the different functional carbon atoms. This in turn
depends on: a) the relative electron withdrawing effects of the
different ........................................X
groups, and on
....................................b)
the degree to which delocalisation of a lone pair from ........................................the
X-group occurs.
Thus
the reactivity of carboxylic acids and their derivatives decreases in
the order:
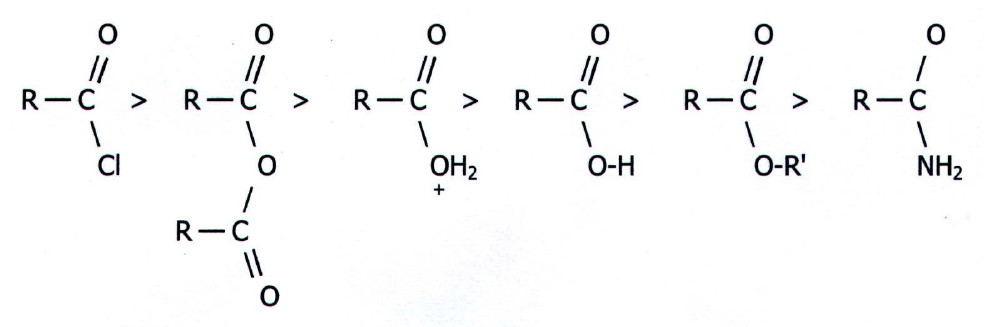
Acid
chlorides are the most reactive because chlorine is a good leaving
group and because delocalisation of a chlorine lone pair is not as
pronounced as it is with oxygen or nitrogen lone pairs. This is
predictable because the chlorine lone pairs are in 3p orbitals which
must overlap with a carbon 2p orbital. With oxygen and nitrogen the
delocalised lone pairs are in 2p orbitals which overlap more easily
with the carbon 2p orbitals.
Anhydrides, the acid itself, and esters, are all more reactive than amides because oxygen is more electronegative than nitrogen. The ester is less reactive than the acid itself due to the electron pushing effect of the R' group in esters.
Finally, acid anhydrides are more reactive than caboxylic acids (and therefore than esters) because the delocalisation of the oxygen lone pairs is shared over two carbonyl groups. This less effectively increases electron density around the functional carbon atoms.
22.3.6. Some nucleophiles which react with carboxylic acids and their derivatives, with the emphasis on illustrating relative reactivities.
i) Water
Acid chlorides react vigorously with cold water and actually fume in moist air. Anhydrides react with cold water, but more slowly - heat is really needed for a reasonable rate of reaction. The hydrolysis of esters requires heating with acid or alkali, as does the hydrolysis of amides. The normal method for hydrolysis of esters or amides is boiling under reflux with either aqueous sulphuric acid or aqueous sodium hydroxide.
In all cases, hydrolysis produces the corresponding carboxylic acid or its anion, depending on pH. The inorganic product depends on the particular derivative reacting, and also on the pH. The pH effect is seen with amides, for example, which give ammonia on alkaline hydrolysis and ammonium ions on acid hydrolysis.
Eg.. RCOCl(l).. +..
H2O(l).... ![]() ....
RCOOH(aq).. +..
H+(aq)..
+..
Cl-(aq)
....
RCOOH(aq).. +..
H+(aq)..
+..
Cl-(aq)
The hydrolysis
of
esters is almost a subject in itself. The most common
mechanisms are variations of two-step nucleophilic substitution
(mechanism 4, FIG. 20.1.), whether carried out in acidic or basic
conditions. However, hydrolysis may occur via mechanisms which involve
fission of the alkyl oxygen bond rather than the acyl oxygen bond:
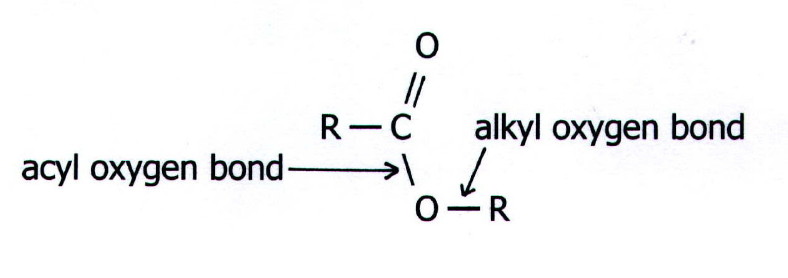
Nevertheless, the most
common mechanism is base "catalysed" (B), bimolecular (2), and involves
acyl oxygen fission (AC) ie BAC2:
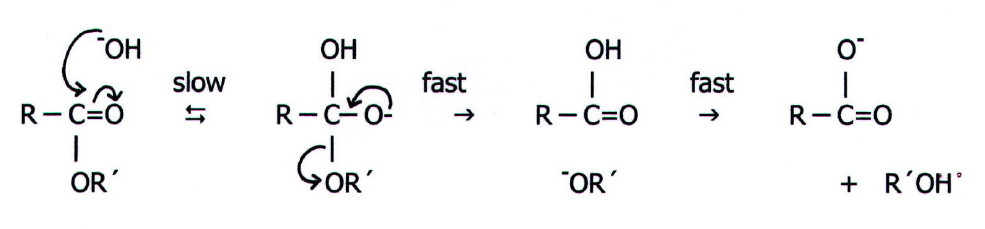
It can be seen that this is simply a version of mechanism 4
(FIG. 20.1.). However, the hydroxide ion is not a
catalyst, it is the nucleophile.
The most common acid
(A) catalysed mechanism
is AAC2. It is also a variation of mechanism 4
and, in addition, is the most common mechanism of esterification. Note
that esterification cannot occur in alkaline conditions because the
carboxylic acid would be dissociated.
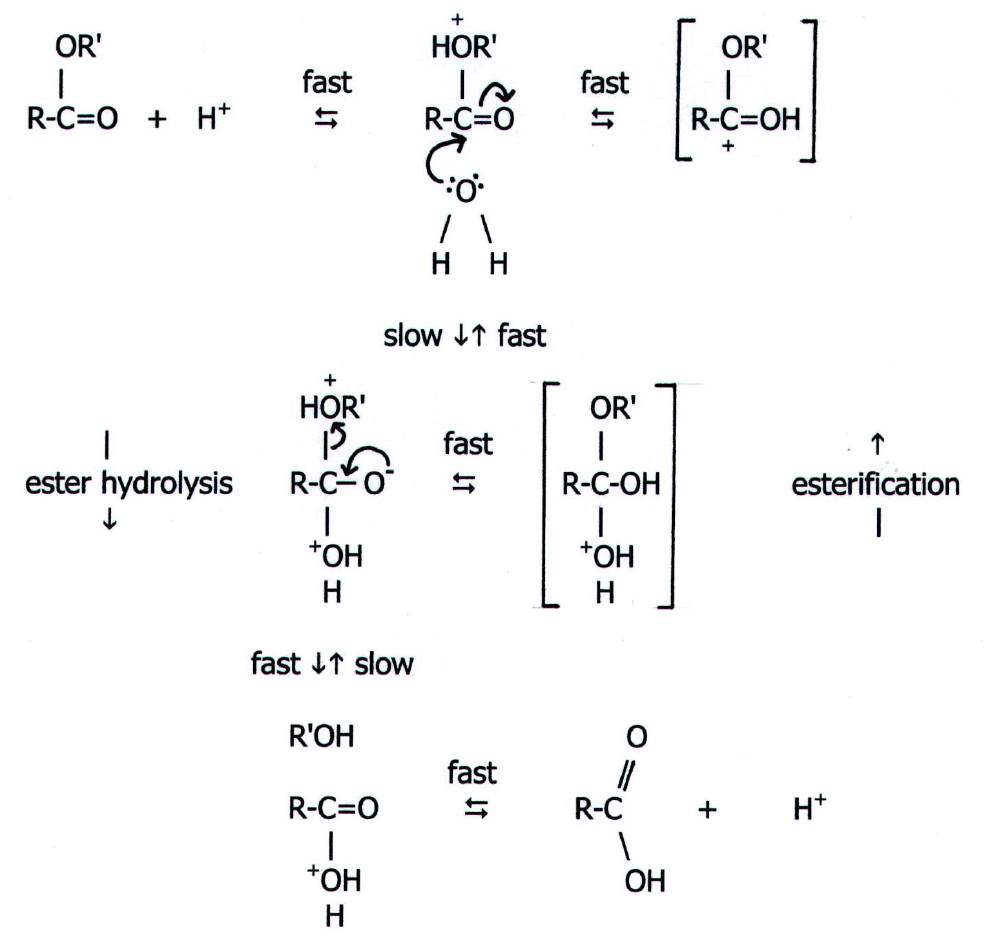
ii) Alcohol
Acid chlorides react readily with alcohols at room temperature. Acid anhydrides require warming. The acid catalysed reaction with carboxylic acids discussed above requires heating under reflux. The reaction of amides with alcohols is insignificant.
In each case the organic product is an ester and the inorganic product obviously varies.
Eg.....
(RCO)2O...
+...
R´OH.... ![]() ....
RCOOH...
+... RCOOR´
....
RCOOH...
+... RCOOR´
Note that with phenol, acid chlorides are the only derivatives to give a useful yield of phenyl esters.
iii) With ammonia and amines
Both acid chlorides and acid anhydrides react at room temperature. To control the reaction and give the best yield of amide, low temperatures and/or a large excess of ammonia solution should be used, especially with the chloride. With esters, it is best to heat with ethanolic ammonia.
Eg.....
RCOOR´(l)...+... NH3(alc).... ![]() ....
RCONH2(s)...
+...
R´OH(l)
....
RCONH2(s)...
+...
R´OH(l)
With carboxylic acids themselves, it is easy to fall into the trap of predicting susceptibility to nucleophilic attack. However, ammonia is a base and undergoes an acid/base reaction with carboxylic acids:
RCOOH(aq)... +...
NH3(aq).... ![]() ....
RCOO-(aq)...
+...
NH4+(aq)
....
RCOO-(aq)...
+...
NH4+(aq)
Carboxylic acids and their derivatives also react with amines in a similar way, though only acid chlorides react significantly with phenylamine.
room temp.
Eg RCOCl(l)..
+..
2R´NH2(l)... ![]() ...
RCONHR´(s).. +..
R´NH3Cl(s)
...
RCONHR´(s).. +..
R´NH3Cl(s)
iv) Carboxylate ions
When distilled with sodium salts of carboxylic acids, acid chlorides produce acid anhydrides:
RCOCl(l)..
+..
R´COONa(s)... ![]() ....RCO.O.CO.R´(l)..
+..
NaCl(s)
....RCO.O.CO.R´(l)..
+..
NaCl(s)
The acid itself may be used instead of the salt, but the yield is not as good, even in the presence of pyridine which is used to remove HCl and pull the equilibrium towards anhydride formation.
v) AlH4- and BH4- ions
The carboxylic acid and all its derivatives (except amides) are readily reduced by the above ions to the corresponding primary alcohols. However, it is impossible to stop the reaction at the aldehyde stage. In contrast with the other derivatives, amides are reduced to the corresponding amine (cf. section 22.3.11.i.). Relative reactivities are hardly relevant.
..........................
LiAlH4/DRY ether
Eg..... RCONH2(s).......... ![]() ..........
RCH2NH2(l)
..........
RCH2NH2(l)
......................
LiAlH4/DRY ether
....... RCOOH(l).......... ![]() ..........
RCH2OH(l)
..........
RCH2OH(l)
......................
followed by H2O
Note that there are other reducing agents which tend to be used for particular derivatives:
acid chloride.. - hydrogen in xylene
solvent with platinum or<
......................
paladium catalyst. If a paladium catalyst poisoned
......................
with barium sulphate and sulphur is used in
......................
quinoline as solvent, the acid chloride is reduced
......................
only
as far as the aldehyde.
esters/amides - sodium in ethanol at room temperature
vi) Halogens
Direct halogenation by halide ions is not possible. The halogenating agents, PCl5, PCl3, and SOCl2 must be used in reaction with the carboxylic acid. In this case, high yields of the corresponding acid chloride are obtained.
Eg...
RCOOH(l)... +... PCl5(s)........ ![]() ........
RCOCl(l)...
+... HCl(g)
........
RCOCl(l)...
+... HCl(g)
Note that the reaction with phosphorus pentachloride indicates the presence of an OH group (cf reaction with alcohols in section 22.1.3.) since HCl is produced, giving steamy white fumes. There is also some interesting theory relating to the choice of halogenating agent in laboratory preparations:
For low boiling point acid chlorides (eg ethanoyl chloride) the best agent is phosphorus trichloride. The chloride is easily separated by distillation from the mixture (H3PO3 decomposes at 200°C)
For high boiling point chlorides (eg bezoyl chloride), phosphorus pentachloride can be used. Fractional distillation gives first POCl3, then the acid chloride.
For preparation of acid chlorides with intermediate boiling points (eg propanoyl chloride) the best agent is sulhpur dichloride oxide. Gaseous sulphur dioxide and hydrogen chloride pass off first.
vii) Benzene
The acylation of benzene by acid chlorides in the presence of halogen carriers has already been discussed in chapter 21, section 21.6.2. However, the emphasis was different: the acid chloride was treated as an electrophile rather than as a molecule undergoing nucleophilic attack.
We did not mention in section 21.6.2. that acid anhydrides also give the reaction.
22.3.7. Predictions iii) Carboxylic acids have available lone pairs on the hydroxyl carbon and might be expected to act as nucleophiles. Delocalisation of the lone pairs over the carboxyl group reduces the availability. However, the carboxylate ion, with its extra lone pair and negative charge, is a better nucleophile. Thus despite the delocalisation, carboxylic acids, and to a greater extent the carboxylate anion, act as nucleophiles.
Amides also act in this way, but examples are insignificant.
Moreover, the same essential reasoning also predicts that carboxylic acids, esters, and amides will act as weak bases.
22.3.8. Examples of nucleophilic and basic activity
The reaction of carboxylic acid salts with acid chlorides in section 22.3.6.iv. is an example of carboxylate ions acting as nucleophiles. The acid itself can react with acid chlorides, but the yield is not as good.
The protonation of carboxylic acids, esters, and amides is an important step in some of the reactions described above, but it is also an example of basic behaviour.
Compared with other
nitrogenous bases, amides are not strong. This is because the lone pair
is delocalised over the functional group as described previously.
Indeed, it is even possible for amides to act as very weak acids with
the resultant anion being stabilised thus:
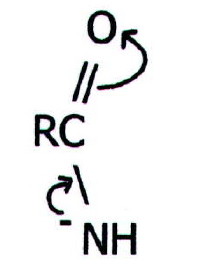
They can sometimes form metal salts containing the above ion.
22.3.9. Other general reactions
i) Halogenation of the alkyl groups
Do not confuse this
with the
reaction of the OH group in carboxylic acids with halogenating agents
(section 22.3.6.v.). Hydrogen atoms in the alkyl chain, particularly in
the -position, are replaced by homolytic
substitution. Chlorine or
bromine in the presence of UV light (or catalysts such as iodine or red
phosphorus) can be used for the reaction:
E.g...
CH3CH2COOH(l). +.
Cl2(g)... ![]() ...
CH3CHClCOOH(l).
+.
HCl(g).. - -
...
CH3CHClCOOH(l).
+.
HCl(g).. - - ![]() etc..
etc..
22.3.10. Other reactions of carboxylic acids and their salts.
i) Heating calcium and other salts
Calcium methanoate gives methanal on heating, whereas the calcium salts of other carboxylic acids give ketones. If calcium methanoate is heated with the calcium salt of another carboxylic acid, the products will be mixed eg calcium methanoate plus calcium ethanoate will give methanal, propanone, and also ethanal.
..........................
400°C
(HCOO)2Ca(s)........
![]() ........
CaCO3(s)..
+..
HCHO(g)
........
CaCO3(s)..
+..
HCHO(g)
..............................
400°C
(CH3COO)2Ca(s)........ ![]() ........
CaCO3(s)..
+..
(CH3)2C=O
........
CaCO3(s)..
+..
(CH3)2C=O
Note that heating carboxylates of other metals, such as sodium, also gives ketones, but alkali metal methanoates give ethanedioates (oxalates) and hydrogen:
.........................
heat
2HCOONa(s)........
![]() ........
Na2C2O4(s).. +..
H2(g)
........
Na2C2O4(s).. +..
H2(g)
Stronger heating will produce carbon monoxide because the ethanedioate breaks down:
...................
strong heat
Na2C2O4(s)........ ![]() ........
Na2CO3(s).. +..
CO(g)
........
Na2CO3(s).. +..
CO(g)
Note that heating ethanedioic acid, itself, to about 150°C produces methanoic acid and carbon dioxide:
HOOC-COOH(l)........ ![]() ........
HCOOH(g)..
+.. CO2(g)
........
HCOOH(g)..
+.. CO2(g)
Benzoates burn on strong heating eg on a peice of porcelain.
ii) Decarboxylation
This is brought about by heating the sodium salt of a carboxylic acid with soda lime (an intimate mixture of sodium hydroxide and calcium oxide):
heat
Eg:..
CH3COONa(s)..
+..
NaOH(from soda lime)... ![]() ...
CH4(g)..+.
Na2CO3(s)
...
CH4(g)..+.
Na2CO3(s)
iii) Heating with sulphuric acid
The release of free acid from salts by heating with concentrated sulphuric acid has already been described in section 22.3.2. However, dilute sulphuric acid must be used to obtain methanoic acid from methanoates, since heating with concentrated sulphuric acid produces carbon monoxide. Given the same treatment, oxalates give both carbon monoxide and carbon dioxide.
.....................................
.. . .....
heat
HCOONa(s) + (conc)H2SO4(l).... ![]() ....
CO(g) + NaHSO4(s/aq) + H2O(l)
....
CO(g) + NaHSO4(s/aq) + H2O(l)
...................................
...... ....
heat
Na2C2O4(s)
+ (conc)H2SO4(l).... ![]() ....
CO(g) + CO2(g) + Na2SO4(s/aq)
+ H2O(l)
....
CO(g) + CO2(g) + Na2SO4(s/aq)
+ H2O(l)
Many of these reactions are useful as distinguishing tests. Note that hydrocarbons burn, hydrogen ignites explosively, carbon monoxide burns with a blue flame, carbon dioxide turns limewater milky, etc. These properties are useful for testing the products of the above reactions.
iv) With iron(III) chloride
In neutral solutions, carboxylates give some useful identifying rections with iron(III) chloride:
Anions of lower carboxylic acids (methanoates, ethanoates) give a red solution (change of iron(III) ligands) followed by a brown precipitate (basic iron(III) salt) on boiling.
Anions of aromatic carboxylic acids give a buff precipitate (a basic iron(III) salt).
Anions of hydroxy-aliphatic acids give a yellow solution (change of iron(III) ligands).
Compare these with the purple solution given by phenols (section 22.1.12.ii.).
v) Carbonyl reactions
We have already seen
that
carboxylic acids do not show carbonyl properties (section 22.3.4).
However, methanoic acid is an exception as can be seen from its
structure:
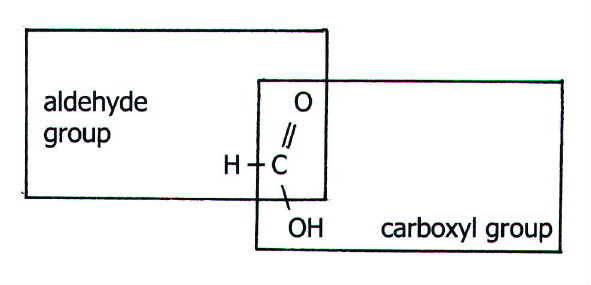
Thus,
methanoic acid is readily oxidised to carbon dioxide and water, and
gives positive tests with Fehling's and Tollen's reagents.
HCOOH..
+.. (O)..... g.....
H2CO3..... ![]() .....
CO2..
+.. H2O
.....
CO2..
+.. H2O
Note that ethanedioates (oxalates) are oxidised by acid manganate(VII) to carbon dioxide, though not by milder oxidising agents.
22.3.11 Other reactions of amides
i) The Hofmann degradation
We shall allow the egocentric name in this case because the reaction has special significance. Amides are reduced to amines with the same number of carbon atoms, but the Hofmann degradation changes them to amines with one less carbon atom.
As part of a reaction pathway, the Hofmann degradation is therefore a useful method of descending a homologous series.
The reagent is bromine dissolved in potassium hydroxide (compare with, but do not confuse with, the bromoform reaction!) and the reaction involves a molecular rearrangement:
RCONH2... +...
Br2..... ![]() .....
RCONHBr... +....HBr
.....
RCONHBr... +....HBr
RCONHBr.. +..
KOH..... ![]() .....
RNCO...+.. KBr..
+.. H2O
.....
RNCO...+.. KBr..
+.. H2O
RNCO....
+.... 2KOH.... ![]() ....
RNH2....
+.... K2CO3
....
RNH2....
+.... K2CO3
The first two steps can occur in the cold, but the third requires heating to 70°C. In practice, the amide is simply heated with the reagent.
ii) Dehydration to give nitriles
Dehydration is acheived by distilling the amide with phosphorus pentoxide
RCONH2(l)... +...
P2O5(s).... ![]() ....
RCN(l)....+....2HPO3(l)
....
RCN(l)....+....2HPO3(l)
iii) With nitrous acid (nitric(III) acid)
Treatment with sodium nitrate(III) dissolved in dilute nitric acid converts amides to the corresponding carboxylic acids (cf the reactions of amines with nitric(III) acid in section 22.4.5.i.).
RCONH2(aq)..+..HONO(aq)... ![]() ...
RCOOH(aq). +.
H2O(l).
+.
N2(g)
...
RCOOH(aq). +.
H2O(l).
+.
N2(g)
22.4.1. Predictions i)
There is an available lone pair on the nitrogen atom in amines, enabling them to act as bases or nucleophiles.
22.4.2. Effect of the rest of the molecule on basic strength (and nucleophilc strength).
There are two main factors which affect the basic strength of amines:
i) Availability and
accessibility of the lone
pair
ii) Ease of solvation of the resultant cation
Thus electron pushing
groups
such as alkyl groups increase the availability of the lone pair and
increase basic strength. However, there is an irregularity in the
following series. It might be expected that base strength would
decrease in the order:
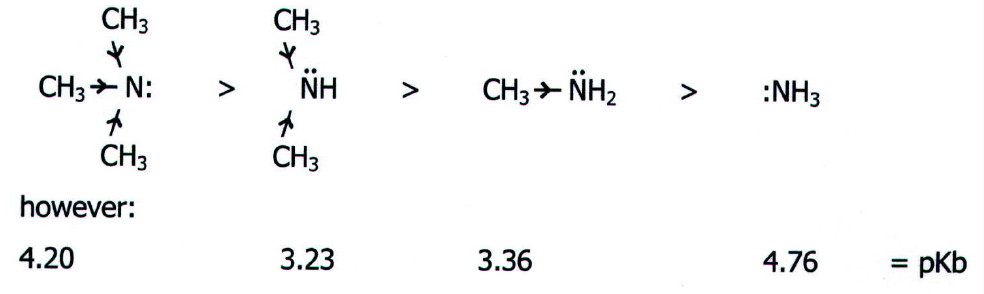
i.e.,
the pKb values show that dimethylamine is hardly more basic than
methylamine, and that trimethylamine is considerably less basic than
methylamine.
This
is because it becomes increasingly more difficult for the resultant
cations to be hydrated. This, in turn, is because the smaller the
number of hydrogen atoms on the cation, the less possible is hydrogen
bonding with water molecules:

Note that quarternary ammonium hydroxides (eg (CH3)4NOH)
are strong bases because they dissociate in aqueous solution to give
hydroxide ions.
Groups
which reduce availability of the lone pair reduce base strength. Thus
phenylamine (aniline) is weaker than ammonia because the lone pair is
delocalised into the benzene ring. Amides are weaker still, because the
lone pair is delocalised onto an electronegative oxygen atom:
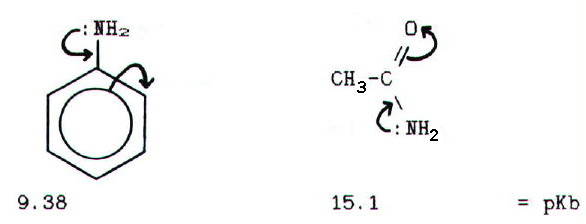
22.4.3. Reactions
as bases
i) Dissociation in water
The concentration of hydroxide ions produced by an amine in aqueous solution is clearly a function of its strength as a base and therefore of the availability of its lone pair:
RNH2(aq)... +...
H-O-H(l)... ...
RNH3+(aq)... +...
-OH(aq)
However, do not forget that it also depends on the solubility of the amine itself.
ii) Salt formation with acids
Primary, secondary, and tertiary amines react with acids to produce salts:
....................... hydrochloric acid.... .......................... salt
RNH2(l)... +...
H+(aq)...
+...
Cl-(aq)... ![]() ...
RNH3+(aq)... +...
Cl-(aq)
...
RNH3+(aq)... +...
Cl-(aq)
R2NH(l)... +...
H+(aq)...
+...
Cl-(aq)... ![]() ...
R2NH2+(aq)... +...
Cl-(aq)
...
R2NH2+(aq)... +...
Cl-(aq)
R3N(l)... +.....
H+(aq)...
+...
Cl-(aq)... ![]() ...
R3NH+(aq)... +...
Cl-(aq)
...
R3NH+(aq)... +...
Cl-(aq)
22.4.4. Reactions as nucleophiles
The reactions of amines as nucleophiles have been covered under previously discussed functional groups. If you have been applying the principle of predicting reactions, you should immediately know where to look them up.
The two important types of compound susceptible to nucleophilic attack by amines are haloalkanes and carboxylic acid derivatives.
i) The reactions of haloalkanes with amines can be seen as part of the sequence of reactions (starting with the reaction between haloalkanes and ammonia) used to prepare amines and, ultimately, quarternary ammonium salts:
..........................
ammonia.............. ![]() ...
primary amine (salt)
...
primary amine (salt)
..........................
primary amine...... ![]() ...
secondary amine (salt)
...
secondary amine (salt)
haloalkane... +
..........................
secondary amine... ![]() ...
tertiary amine (salt)
...
tertiary amine (salt)
..........................
tertiary amine....... ![]() ...
quarternary ammonium salt.
...
quarternary ammonium salt.
To release free amines from the salts initially produced, it is necessary to treat the reaction mixture with alkali.
However, this is not a very useful method for preparing a particular class of amine, because it is difficult to avoid a mixture of products. The yield of primary amine in the first reaction can be increased by starting with an excess of ammonia. The yield of quarternary ammonium compound can be increased by using excess haloalkane.
ii) The reactions of carboxylic acid derivatives (particularly acid chlorides and anhydrides) with amines produces N-substituted amides:
.................................
primary amine......![]() ...
N-alkyl acid amide
...
N-alkyl acid amide
acid chloride
........................ +..... secondary amine... ![]() ...
N,N-dialkyl acid amide
...
N,N-dialkyl acid amide
or anhydride
.................................
tertiary amine....... ![]() ...
salt (cf reaction with haloalkanes)
...
salt (cf reaction with haloalkanes)
iii) With aldehydes and ketones See section 22.2.2.
22.4.4. Other reactions of amines
i) With nitric(III) (nitrous) acid
The reagent is sodium nitrate(III) and dilute hydrochloric or sulphuric acid, and the reactions are useful as tests and in preparations.
| Table 22.5. Summary of the reactions of amines with nitric (III) (nitrous) acid. | ||
| type of amine: | Aliphatic | Aromatic |
| primary | vigorously evolved (test). Note, yield of corresponding alcohol sometimes excellent, sometimes bad. |
Between
0
and 5°C give diazonium salts ArN2+X-. At higher temperatures, nitrogen is evolved and phenols produced. |
| secondary | Nitrosation |
|
| tertiary | No reaction apart from dissolving in the acid. |
Electrophilc
substitution of -NO into activated benzene ring, mainly in 4 position. |
Notes:
Note i)
Note that the production of alcohols in the reaction with primary
aliphatic amines occurs via the diazonium ion:
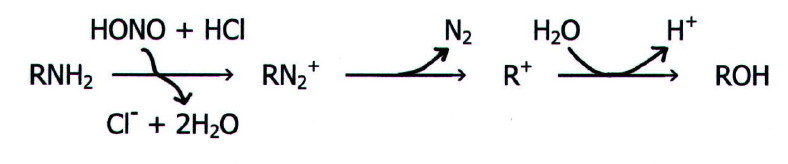
This accounts for the variety of products sometimes obtained:
the carbonium ion does not react only with water. In fact,
1-aminopropane gives propan-1-ol in only about 7% yield. Propan-2-ol
and propene are also formed. Moreover, when hydrochloric acid is used
rather than sulphuric(VI) acid, 1- and 2-chloropropane are also formed.
With methylamine, the main product is methyl nitrate(III) (nitrite), but ethylamine and other amines give an excellent yield. Thus, in combination with the Hofmann degradation (section 22.3.11.i.), this reaction is often used as part of a pathway to descend a homologous series:
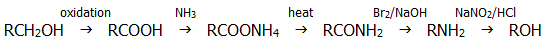
Note ii) The importance of diazonium compounds is shown in section 22.5.
Note iii) The nitrosation of secondary amines involves the nitronium ion:
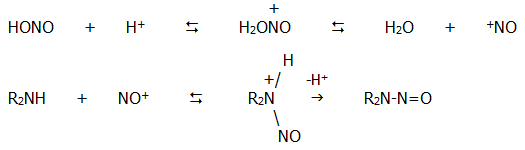
ii) Nuclear reactions of phenylamine (aniline)
The nitrosation of phenylamine shown in table 22.5. is just one example of electrophilic substitution into phenylamine's activated benzene ring.
With bromine water,
phenylamine gives a faily instant white precipitate of
2,4,6-tribromophenylamine at room temperature.
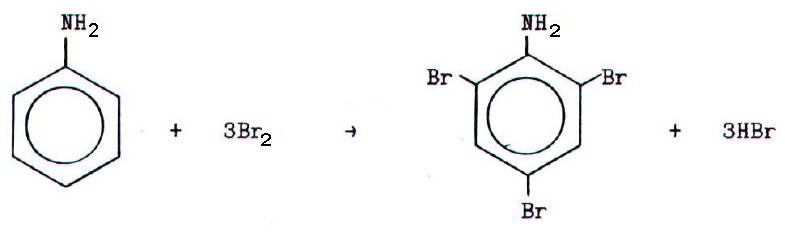
Nitration with concentrated nitric acid(V) dissolved in
concentrated sulphuric(VI) acid, and acylation in the presence of a
halogen carrier are also possible. However, the amino group must first
be protected.
This
is done by acetylating the amino group using ethanoyl anhydride, and
then hydrolysing the protected group to restore the amino group at the
end of the reaction:
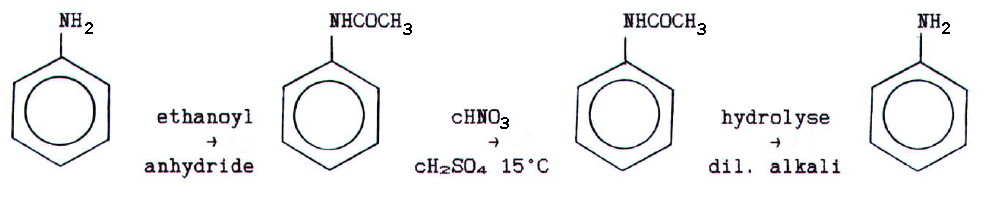
Note
that nitric acid would oxidise the amino group and that in the
acylation reaction, it is the halogen carrier which would react with
the amino group.
22.5.1. The great importance of diazonium compounds stems from three facts:
i) Industrially, they are used to make diazo dyes. The diazonium ion acts as a nucleophile.
ii) Theoretically, they allow the benzene ring to undergo nucleophilic substitution.
iii) Practically their susceptibility to nucleophilic substitution makes them useful for a large number of laboratory preparations.
Note that if something is theoretically possible, it is also possible in practice (unless the theory is wrong e.g. some additional factor has not been considered).
22.5.2. Diazo
dyes
are made by coupling diazoniums with activated
benzene rings. The simplest examples of coupling are with phenol and
phenylamine:
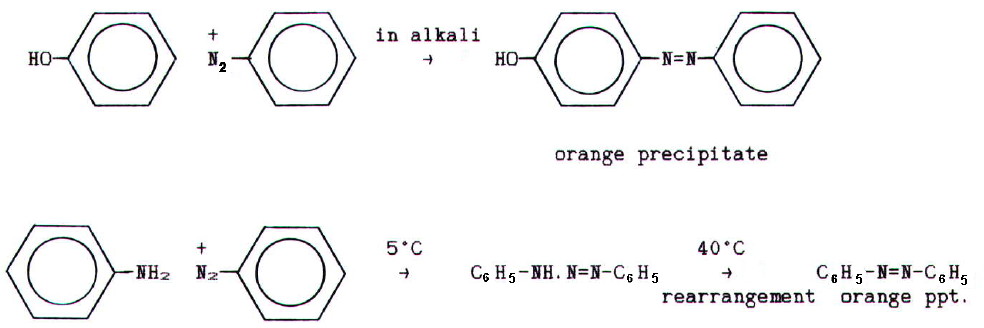
Industrial diazo dyes are more complex than this. Even in the
laboratory it is substances like napthalen-2-ol (b-napthol)
that are used to test for primary aryl amines:
- The test substance is mixed in a test tube with aqueous sodium nitrate(III) (nitrite) solution and cooled in an ice/water bath to 5°C.
- A solution of dilute hydrochloric or sulphuric(VI) acid is similarly cooled.
- The acid is added slowly to the test mixture, maintaining the temperature at 5°C.
- Napthalen-2-ol solution is added carefully to the reaction mixture
- If a primary aryl amine is present, a red precipitate of diazo dye will form.
22.5.3. Nucleophilic attack on diazoniums
The structure of the
diazonium ion is
somewhere between the two structures shown below:
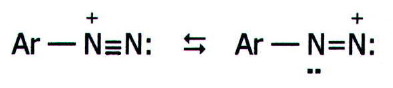
It is the effectiveness of nitrogen as a leaving group which
makes these extraordinary nucleophilic reactions possible. The
mechanism is regarded as SN1, with the
intermediate formation of an aryl carbonium ion!

The host of nucleophiles which can react with the aryl
carbonium ion is summarised in the table below:
| TABLE 22.6. Nucleophiles which react with diazonium compounds. | ||
| Nucleophile | Reagent/Conditions | Main organic product |
| H2O | Dilute acid/boil | Ar-OH |
| "F-" | HBF4/heat | Ar-F |
| Cl- | CuCl | Ar-Cl |
| Br- | CuBr | Ar-Br |
| I- | KI | Ar-I |
| -CN | KCN | Ar-CN |
| "H+" | H3PO2 | Ar-H |
The value of these reactions in preparative chemistry can not be over-emphasised. All these groups can be introduced into benzene itself via the following pathway:
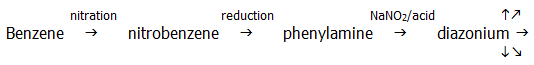
The two important reactions of nitriles are hydrolysis and reduction. Both reactions are useful in sequences used to ascend a homologous series.
i) Hydrolysis is important as the second step for introducing carboxyl groups into a compound. However, the free acid is not immediately formed:
........
acid hydrolysis
RCN........... ![]() ............
RCOO-(aq)..... +..... NH4+(aq)
............
RCOO-(aq)..... +..... NH4+(aq)
......
alkaline hydrolysis
RCN........... ![]() ............
RCOO-(aq)..... +..... M+(aq)
............
RCOO-(aq)..... +..... M+(aq)
ii) Reduction (eg with sodium in ethanol) is useful in ascending a homologous series when an amine's further reactions are more useful than those of the carboxylate group:
.............................
(Na/EtOH)
RCN.....
+.....
4[H]........ ![]() .........
RCH2NH2
.........
RCH2NH2
The most important reaction of the nitro group is reduction to a primary amino-group. A common reducing agent is tin in concentrated hydrochloric acid:
........................................
(Sn/HCl)
R/Ar-NO2.....
+.....
6[H]........ ![]() .........
R/Ar-NH2.....
+.....
H2O
.........
R/Ar-NH2.....
+.....
H2O
This is clearly most useful for the preparation of aromatic amines via nitration of the benzene ring, and hence also for the syntheses of benzene derivatives via diazonium compounds (section 22.5.3.).
22.8. QUESTIONS
1) Ammonia nucleophilically attacks haloalkanes and replaces the halogen atom, but does not react similarly with alcohols. How could you have predicted both these reactions? How does ammonia react with phenol?
2) i. What observable differences in the reactions of 1°, 2°, and 3° alcohols with acidified dichromate(VI) enables the reactions to be used as distinguishing tests? ii. Why is it not sensible to add Tollens reagent (silver mirror test) to any of the above reaction mixtures in order to test for the presence of an aldehyde? iii. What additional step would be needed if you wished to use Tollen's reagent to test for aldehydes in the above reaction mixtures?
3) Describe the usefulness of the haloform reaction as a qualitative test. What is it about the iodoform reaction which makes it an even more useful distinguishing test than the other haloform reactions?
4) How do observable differences amongst the reactions of various carbonyls provide information about their structures?
5) Describe how observable differences in the oxidation reactions of alcohols, aldehydes and ketones provide a basis for distinguishing tests.
6) What is the oxidation state of the functional carbon atom in alcohols, aldehydes, ketones, and carboxylic acids respectively (section 11.4.)? Use this information to write balanced equations for the reactions with acidified dichromate(VI) (section 11.5.).
7)
The table below summarises the results of some
tests on a selection of colourless organic liquids A ![]() E (some may be in aqueous
solution). Deduce as much as you can about the structure of each
compound, explaining how you have used the test information in your
reasoning. For each compound discuss one test after another in the most
logical order.
E (some may be in aqueous
solution). Deduce as much as you can about the structure of each
compound, explaining how you have used the test information in your
reasoning. For each compound discuss one test after another in the most
logical order.
| warm with Cr2O72- | heat with alkali | treat with I2/NaOH | treat with PCl5 | |
| A | orange |
no observable change | reaction, no fumes | |
| B | orange |
yellow precipitate | reaction, no fumes | |
| C | no observable change | no observable change | no observable change | white fumes |
| D | no observable change | yellow precipitate | reaction, no fumes | |
| E | no observable change | no observable change | no observable change | white fumes |
If you were recording the above observations in an examination practical, how would you improve the descriptions (section A1.4.4.)
8) Give the theoretical basis underlying the order of acid strength of fluoro-, chloro-, bromo-, and iodoethanoic acids (section 22.3.3.ii.).
9) Explain in more relevant terms the meaning of the phrase "powerful reducing qualities" in section 22.3.4., paragraph 4.
10) Explaining your reasoning, fit protonated esters and protonated amides into the relative reactivity scheme shown in section 22.3.5.
11) Which of the explanations for relative reactivities of carboxylic acid derivatives are kinetic, and which are thermodynamic?
12) How would you ensure the maximum yield of ester from the reaction indicated by the equation in section 22.3.6.ii.?
13) Why would it not be sensible to use dilute aqueous ammonia in the preparation of amides from acid chlorides?
14) What easy evidence could be obtained for the evolution of a hydrocarbon when sodium ethanoate is heated with soda lime? What difference would you see in your test for hydrocarbon if sodium benzoate were heated with soda-lime?
15) How can the reactions described in section 22.3.10. be used to distinguish between different carboxylates?
16) In what ways do the reactions of methanoates differ from those of other aliphatic carboxylates and from those of ethanedioates?
17) Write an overall balanced equation for the Hofmann degradation.
18) What role does nitrogen's lone pair play in the reactions of amines? How is its role affected by the rest of the molecule?
19)
Summarise the importance of nitric(III) and nitric(V) acids in organic
chemistry.
Unless otherwise stated, all materials in this web version of chapter
22 are © 2007 Adrian Faiers MA (Oxon) MCIPR

What
's the connection between a dozen eggs and a garden mole?

Answer: Not a lot, really,
but see Chapter
1