Recommended by:
Top
20 UK science resources,
The Tutor Website
Recommended by:

Rated:

2010
Listed on the science,
engineering
and technology section of 
'providing you with access
to the very best Web resources for education and research, evaluated
and selected by a network of subject specialists.'
(Please note that intute closed in July 2011)
Section 1: Essentials (physical chemistry)
CHAPTER 9: MORE ON
KINETICS
NB This chapter has now been
updated to improve browser compatibility.
Please
use the 'send email' link at the top right hand corner of this page to
report any problems.
QUICK SKIPS:
(Click on the 'back' arrow to get back to this quick skip section)
Determining order
Half life
Reaction
mechanisms from rate expressions, including the molecularity of a
reaction
Molcularity vs.
order
Collision
theory
Transition
state theory
The effects of
temperature, pressure and concentration on rate
Catalysts, their
effect on rate and their mechanisms
The effect of light on rate
9.1. DETERMINING ORDER
In this section we shall look at the two methods of determining order mentioned at the end of section 8.1. We shall look at the methods as applied to a hypothetical reaction:
A
+ 2B ![]() C + 2D
C + 2D
9.1.1. The first method is the more obvious. It finds directly the relationship between initial rate and initial concentration.
Several experiments are performed. In each experiment the starting concentration of one reactant (eg B) is the same, but the starting concentration of the other (in this case A) is varied. Any variation between experiments of the initial rate will depend on the variation in concentration of A. Whatever the number of reactants, the concentration of only one reactant is varied between experiments.
The reaction is followed by measuring the concentration of one reactant or product at various times during the reaction. Alternatively, a continuous method such as spectroscopy can be used. Methods include:
- using a volumetric pipette to take samples from the reaction mixture at measured times, quenching the reaction and titrating for a chosen reactant or product;
- measuring the appearance or disappearance of a coloured reactant or product using a spectrometer / colourimeter;
- measuring
the production of a gas using either a gas syringe or collecting the
gas over water in an inverted, graduated cylinder.
You will be familiar with the other techniques for measuring mass, concentrations, volumes of solutions etc. from your practical classes.
The measured
concentration of one reactant or product is then plotted against time
(FIG. 9.1.). The gradient of the tangent to the start of each curve
then gives the initial rate in each experiment, as shown in FIG. 9.1.
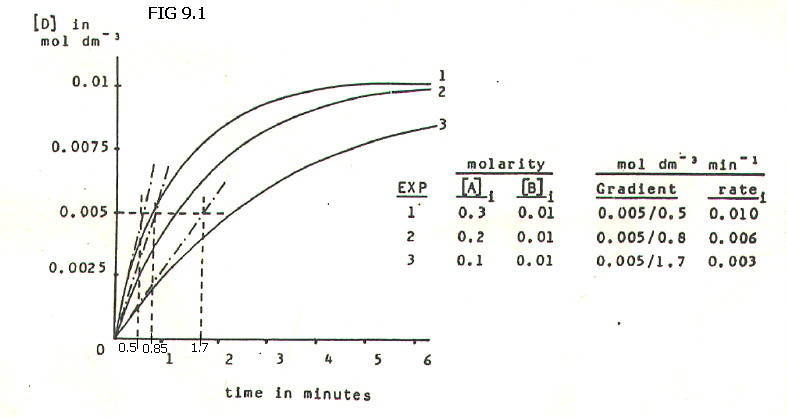
Bearing in mind experimental
error, the following patterns can be seen in the above example. When
the initial concentration of A is doubled from 0.1M to 0.2M, the
initial rate doubles. When the initial concentration is trebled from
0.1M to 0.3M, the initial rate trebles. It can therefore be assumed
that for this reaction initial rate is proportional to initial
concentration of A, and that more generally:
rate 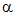 [A] in this
reaction.
[A] in this
reaction.
i.e. the reaction is first order with respect to A.
With less obvious data, it may be necessary to make plots of initial rate against initial concentration, initial concentration squared, initial concentration cubed, and so on. The plot which gives a straight line graph more clearly demonstrates the correct relationship.
9.1.2. The half-life method depends on the relationship:
T˝
1/cin-1
where: T˝ = half-life, the time taken for the reaction to get half way to completion, e.g. half the limiting reactant is used up.
ci = initial concentration of the rate determining reactant
n = order with respect to the rate determining reactant.
In combination with the half-life method, the results from experiment 1 in FIG. 9.1. can be used again, this time to find the order with respect to B.
In experiment 1, the initial concentration of A is 30x higher than that of B. Therefore, even when the reaction is complete, the concentration of A will still be 0.295M. It follows that any significant change in rate during the reaction will be due to changing concentration of B. i.e. B is what we have called the rate-determining reactant, and any change in T˝ will depend on [B]i.
(Note that at this stage "rate-determining" is not a perfect term for B: for all we know, the reaction may turn out to be zero order with respect to B.)
The results of experiment 1 are re-plotted in FIG. 9.2
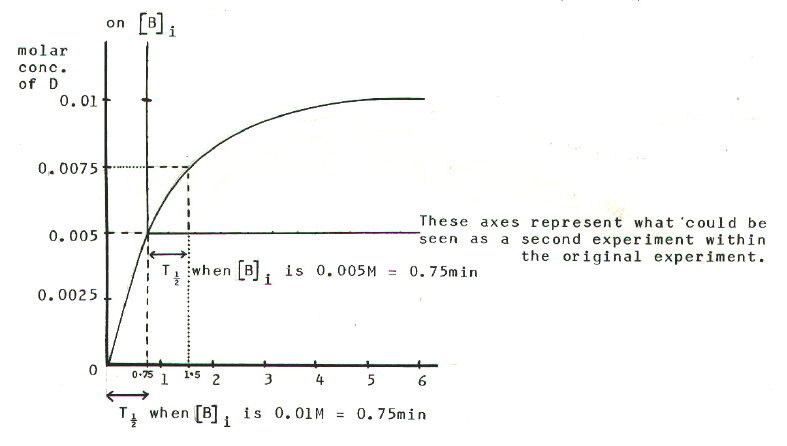
The value of T˝
taken from the second set of axes is the time taken for the
concentration of D to rise from 0.005M to 0.0075M, i.e. half
way between the new starting point and the final value.
Several similar measurements can be made from different starting points
on the graph, in order to clarify any pattern in T˝.
The two values from FIG. 9.2. show clearly that the half-life is
constant.
i.e. T˝
1/ci0
thus n must = 1
i.e. cin-1 = ci1-1 = ci0
In other words, this hypothetical reaction is first order with respect to B.
i.e. rate
[B]
and the overall rate expression for this reaction is:
rate = k[A][B]
Note that this is an example of the order being different from the number of moles of B shown to react by the stoichiometric equation. Moreover, although this is a hypothetical reaction, the same kinetics would be observed if A were S2O82-(aq) and B were I-(aq).
S2O82-(aq)
+ 2I-(aq) ![]() I2(aq) + 2SO42-(aq)
I2(aq) + 2SO42-(aq)
N.B. I2(aq)
+ I-(aq) ![]() I3-(aq)
I3-(aq)
The relationships between T˝ and initial concentration of the "Rate-determining" reactant are summarised in TABLE 9.1.:
|
TABLE 9.1. |
|
|
ORDER |
T˝
is |
|
zero |
ci |
|
first |
independent |
|
second |
1/ci |
|
third |
1/ci2 |
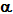
When the data are less clear than those from "experiment 1", T˝ should be plotted against the various functions of ci shown in the right hand column. The plot producing a straight line graph clearly demonstrates the correct relationship and the order can thus be deduced.
9.2. REACTION MECHANISMS FROM RATE EQUATIONS
9.2.1. We have now seen that order does not always equal the number of moles shown reacting in the stoichiometric equation for a reaction. This suggests that reactions might not always proceed in the way we would expect.
For example, from the
equation A + 2B ![]() C + 2D, we might predict that one particle of
A and two of B collide, react, and give products. This would be a trimolecular
reaction, i.e. the molecularity = 3.
C + 2D, we might predict that one particle of
A and two of B collide, react, and give products. This would be a trimolecular
reaction, i.e. the molecularity = 3.
9.2.2. The molecularity of a reaction step is the number of reactant particles which must collide for that step to occur.
9.2.3. The overall molecularity of a reaction is the molecularity of the step which determines the overall rate i.e. the slowest step, known as the rate determining step.
9.2.4. Returning to the example in section 9.2.1., the first thing to say is that trimolecular reactions are rare. Moreover, if two particles of B were involved in the rate-determining step, the rate might be expected to depend on [B]2.
It is quite possible for the rate to be proportional to [A][B] (section 9.1.). Such a relationship would suggest that only one particle of A and one of B collide and react in the rate determining step. The molecularity in this case is 2, a bimolecular reaction.
We can go on to suggest sequences of reaction steps that would be consistent with this:
FIRST SUGGESTION:
A + B ![]() C + X...............
slow
C + X...............
slow
X + B ![]() 2D....................
fast
2D....................
fast
SECOND SUGGESTION:
A + B ![]() D + Y...............
slow
D + Y...............
slow
B + Y ![]() C + D...............
fast
C + D...............
fast
THIRD SUGGESTION:
A + B ![]() *AB.................
slow
*AB.................
slow
*AB + B ![]() C + 2D.........
fast
C + 2D.........
fast
These sequences of reaction steps are known as reaction mechanisms. They can be quite detailed, as we shall see in the organic section of this book. Even more experiments must be performed in order to determine which, if any, of the mechanisms suggested above is correct. For example, if the presence of X could be demonstrated during the reaction between A and B, this would be evidence consistent with the first suggestion.
9.3. MOLECULARITY VS. ORDER
9.3.1. Just as order does not necessarily equal the number of moles in the stoichiometric equation, so molecularity does not always equal order. Here are three examples.
i) In experiment 1 (section 9.1.), A was present in such high concentration that the changing rate during the course of the experiment depended only on the changing concentration of B, i.e. rate = k[B], a first order relationship. The reaction thus showed, so-called, pseudo first order kinetics, despite occurring via a bimolecular mechanism.
ii) When a secondary haloalkane undergoes hydrolysis in certain mixtures of potassium hydroxide, alcohol, and water, substitution may simultaneously occur via unimolecular and bimolecular mechanisms, SN1 and SN2 (sections 20.4. and 20.5.). It will therefore simultaneously follow first order and second order kinetics, and the order will be fractional, somewhere between 1 and 2.
iii) If you think the two previous examples are cheats, here is a good one:
The reaction 2A +
B ![]() C + D may occur via the following two bimolecular
steps:
C + D may occur via the following two bimolecular
steps:
A + B  X...............
fast
X...............
fast
A + X ![]() C + D........
slow
C + D........
slow
in this case:
rate
[A][X]
i.e.
[A][A][B]
i.e. [A]2[B]
9.4. COLLISION THEORY
9.4.1. The reaction mechanisms above are based on two models of the interactions between reactant particles:
1) Collision theory.
2) Transition state theory
The models overlap in many respects, but there are key differences.
9.4.2. Collision theory is based on the following idea: in order for a reaction to occur, reactant particles must collide with a certain minimum kinetic energy, the activation energy, or Eact, or Ea. The value of the activation energy depends on the particular reaction.
We have already seen that the kinetic energy of reactant particles is a function of temperature. In section 5.4.9., we saw how the distribution of energies in a gas is a function of the Maxwell-Boltzmann distribution of speeds. The distribution is similar in liquids, and we shall see its uses in sections 9.7. to 9.10.
9.5. TRANSITION STATE THEORY
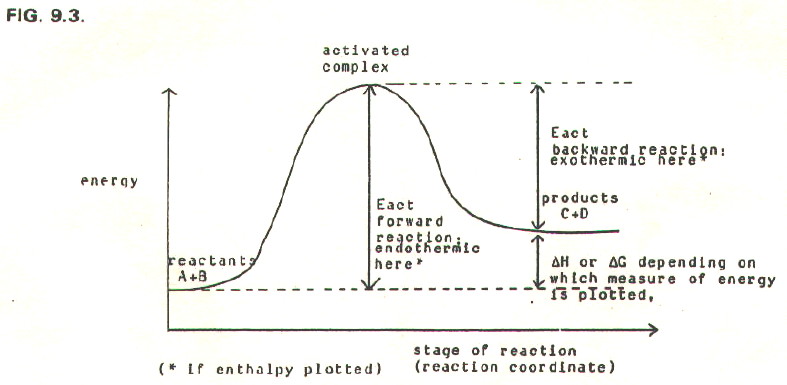
9.5.1. Transition state theory is based on the idea that products and reactants in a reaction (or reaction step) are in equilibrium with an intermediate "activated complex" or "transition state".
FIG. 9.3. plots the
course of a simple bimolecular reaction in terms of transition state
theory. Such diagrams are called reaction profiles.
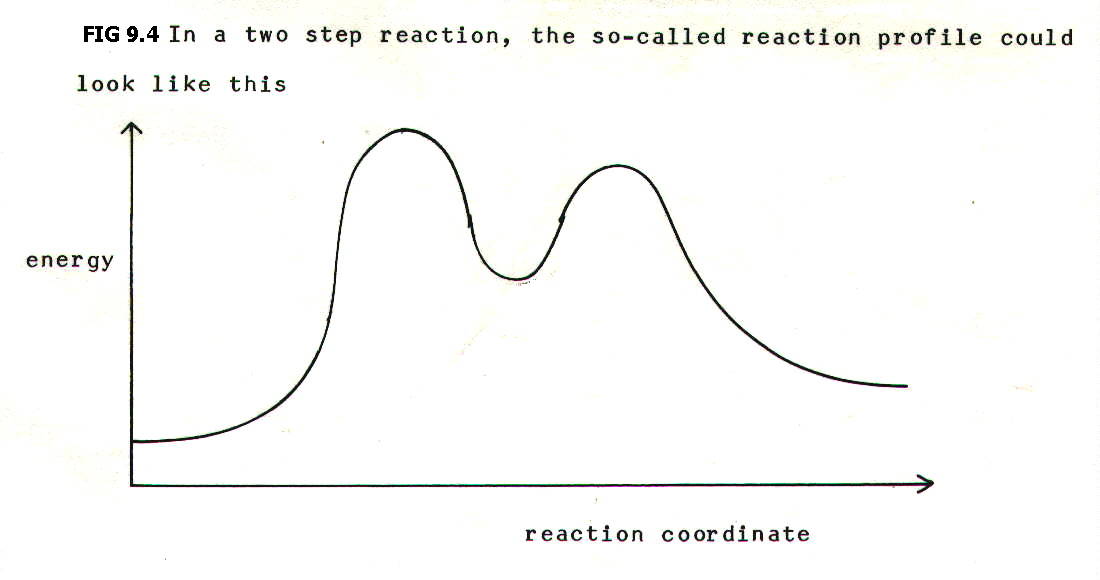
FIG. 9.4. shows the reaction profile for a two step reaction.
9.6. COLLISION VS.
TRANSITION
9.6.1. As usual, neither model should be considered as right or wrong. Each emphasises different points. The key difference between the approaches is that collision theory tackles kinetics via energetics, and transition state theory tackles it via equilibrium and DG (chapters 10 and 12). An understanding which incorporates aspects of both models will often be more useful than sticking rigidly with one or the other.
The collision model is certainly easier to picture and to relate to the changing forces of electrostatic attraction (breaking and making bonds) which occur in reactions.
9.6.2. A combined model might have collisions with sufficient energy producing the transition state i.e. colliding with sufficient vigour to overcome electrostatic forces of attraction (bonds) which must be broken in the reactants. Collision theory does not deny the existence of transition states nor the consideration of energy profiles. Equally, transition state theory does not deny the importance of collisions.
In discussing the effects of, temperature, pressure, concentration and catalysts on reaction rates we shall shamelessly mix the language of both theories (models). In TABLE 8.1. we summarised the effects of these external factors. Now we shall look at them in more detail.
9.7. THE EFFECT OF TEMPERATURE ON RATE
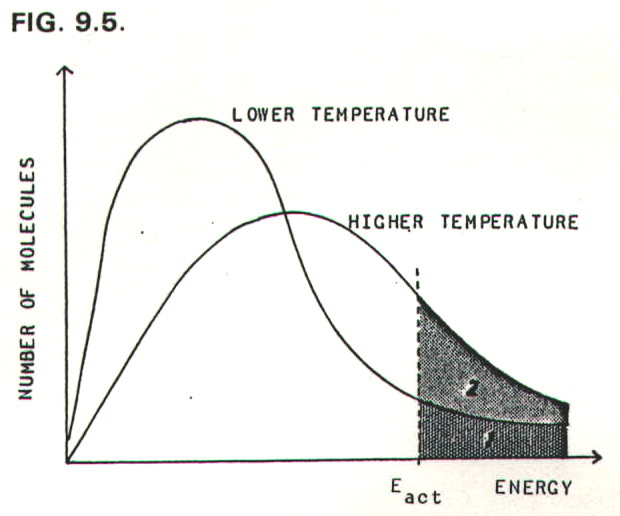
FIG. 9.5. represents the energy
distribution of molecules in a gaseous reaction mixture at two
different temperatures. The activation energy for the reaction is
marked on the graph, and the shaded areas represent the numbers of
molecules with equal to, or greater than, the activation energy.
At the lower temperature the number of molecules with equal to or greater than the activation energy is given by area 1. At the higher temperature the number is greater, and given by (area 1 + area 2).
It therefore follows that the reaction will be faster at the higher temperature since the frequency of collisions with equal to or greater than the activation energy will be higher. There is a minor additional effect: the molecules move more quickly and collide more often at higher temperatures.
The equation for the graphs in FIG. 9.5. is:
N = Noe-E/RT
where N is the number of molecules with energy E in a total population of N0 molecules, R is the universal gas constant, and T is the absolute temperature.
The rate constant for the reaction will be a function of the number of molecules with > or = Ea, and is given by:
k = Ae-Ea/RT
This is the Arrhenius equation and A is the Arrhenius constant. We have already said that not all collisions with sufficient energy result in reaction. For example, particles may collide with the wrong orientation. Such factors are included in the Arrhenius constant.
9.8. THE EFFECT OF PRESSURE ON RATE
This is mainly relevant to gaseous reactions. Increasing the pressure will bring reactant particles closer together. They will therefore collide more frequently and the frequency of favourable collisions will consequently increase. This, in turn, will lead to an increased reaction rate. Decreasing the pressure will have the opposite effects.
9.9. THE EFFECT OF CONCENTRATION ON RATE
Increasing the concentration of a reactant involved in the rate determining step will increase the frequency of relevant collisions. The frequency of successful collisions will correspondingly increase and hence, so too, will the rate. Decreasing the concentration will have the opposite effects. Note that changing the concentration of a reactant not involved in the r.d.s. will not affect the rate.
9.10. THE EFFECT OF CATALYSTS ON RATE
9.10.1. Catalysts are substances which alter the rate of a reaction without themselves being used up. Positive catalysts increase the rate, and negative catalysts (inhibitors) decrease the rate. There are other things which need to be made clear about catalysts:
i) They do not affect the position of equilibrium. They affect the rate at which it is attained, and they affect the rate of both the forward and backward reactions at equilibrium, but they do not affect the composition of the equilibrium mixture.
ii) Often they are effective in small quantities, but this is not always the case. Fairly large amounts of aluminium chloride are needed to catalyse the acylation of benzene by ethanoyl chloride.
iii) At the end of a reaction catalysts are found to be unaltered chemically. This does not mean they play no part in the reaction. They play an active role and may undergo fundamental chemical changes during the course of reaction. They are restored to their original chemical form at the end, but may have changed physically e.g. lumps may have broken down into powder.
iv) Some catalysts are extremely specific. This is particularly true of enzymes which are naturally occurring catalysts.
v) Some catalysts require the presence of other substances called promoters to improve their effectiveness. In addition, most can have their effectiveness reduced or destroyed by poisons. (N.B. Poisons in this context are sometimes referred to as inhibitors, a term also (confusingly) used for negative catalysts.)
9.10.2. The
term catalyst most commonly refers to positive catalysts.
Positive catalysts work by providing an alternative reaction mechanism
with a lower activation energy. Thus in the presence of the catalyst, a
higher proportion of the reactant particles have sufficient energy to
form the intermediate.
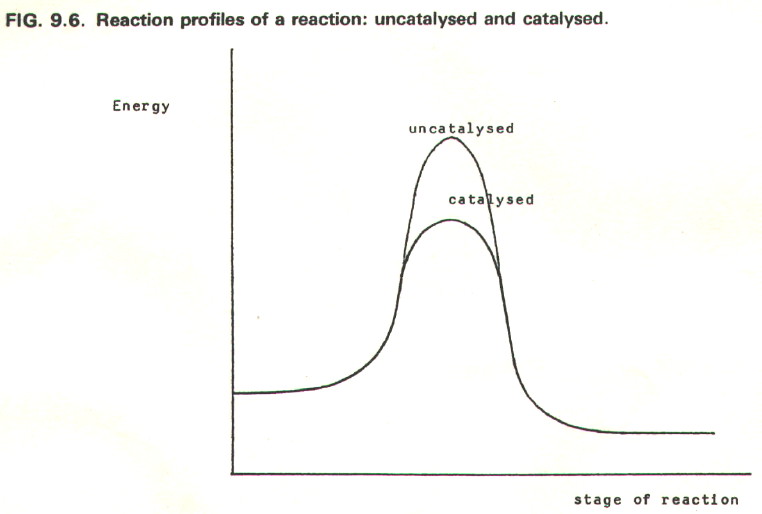
In terms of collision theory,
the frequency of collisions with equal to or greater than the
activation energy increases. Consequently the rate increases. However,
collision theory is not particularly appropriate to heterogeneous
catalysis (section 9.10.3b.).
9.10.3. Catalysts may be in the same phase as the reactants, or in a different phase. These two types of catalysis are given names:
9.10.3a. In homogeneous catalysis, the catalyst is in the same phase as the reactants. For example, in aqueous solution Fe3+ ions catalyse the oxidation of I- ions by S2O82-.
9.10.3b. Heterogeneous catalysts are in a different phase from the reactants. For example, finely divided nickel (solid) catalyses the reaction between hydrogen gas and ethene gas.
9.10.5. Two mechanisms of catalytic action have been suggested. The first, intermediate compound formation, is more appropriate to homogeneous catalysis. The second, adsorption theory, is more appropriate to heterogeneous catalysis.
9.10.5a. The intermediate "compound" in the above example of homogeneous catalysis (section 9.10.3a.) is the Fe2+(aq) ion.
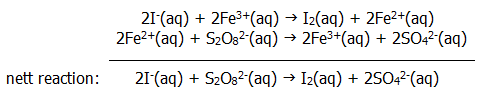
The essential features of the intermediate compound mechanism are demonstrated clearly by this example:
i) The iron(III) ions participate in the reaction, providing a totally different pathway.
ii) Both steps in the catalysed mechanism have a lower activation energy than the uncatalysed reaction.
Interestingly, aqueous chromium(III) ions do not catalyse the reaction. This is because they do not attract another electron and form aqueous chromium(II) ions as readily as iron(III) attracts an extra electron. Can you explain this in view of the relative positions of iron and chromium in the periodic table? (section 18.3.3.).
9.10.5b. Adsorption theory attaches importance to adsorption of the reactants onto the catalyst surface. As well as being appropriate to heterogeneous catalysts, it is also useful when considering enzymes. Enzymes may be in the same phase as the reactants, but their molecules are large, and binding of reactants to an enzyme's "active site" is fundamental to the catalysis.
In the example of heterogeneous catalysis above (section 9.10.4b) the adsorption of hydrogen onto the nickel surface is fundamental. The binding sites on the nickel surface for hydrogen atoms are slightly further apart than the H2 bond length.
This strains the H-H
bond tending to split the molecule into more reactive hydrogen atoms.
The ethene is also adsorbed onto the nickel surface and in such a way
that reaction with the hydrogen "atoms" is encouraged.
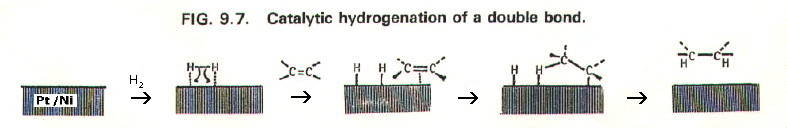
The mechanism is complex, and
such catalysts are often very specific. This is because each of the
many bond making and breaking steps must be faster than the slowest
step in the uncatalysed reaction. Catalysts are often discovered by
trial and error.
9.11. THE EFFECT OF LIGHT ON RATE
9.11.1. Light (especially high energy ultra violet light) can affect the rate of some reactions. It can do so by providing an initial input of energy which may lead to the breaking of bonds, thus making possible an alternative pathway with lower activation energies.
Most usually, light provides reactive free radicals by homolytic bond fission. Often the production of free radicals results in a chain reaction (eg chlorination of methane, section 20.3.).
It is worth noting that
after the initial light activated step, all steps in a chain reaction
are exothermic (or close to exothermic) as well as being fast. Thus
methane does not react with bromine under the same conditions as it
reacts with chlorine. A clue to understanding this comes from the
observation that  H = +93 kJ mol-1 for
the step:
H = +93 kJ mol-1 for
the step:
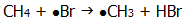
Because bromine's outer shell is further ("and more screened from") the nucleus, the nucleus is less able to attract an electron from a C-H bond into an H-Br bond.
9.12. INTEGRATED RATE EQUATIONS
The half-life method (section 9.1.2.) is based on a relationship which comes from integrating rate equations. If you are unlucky, your examiners may want you to know more about integrated forms of rate equations, in which case you will have to look them up elsewhere.
It is rare for a student's understanding of kinetics at this level to be increased by studying integrated rate equations.
9.13. QUESTIONS
1) How could you follow the reaction between S2O82- ions and I- ions?
2) Suggest possible mechanisms for the reaction between S2O82- ions and I- ions. How could you test your suggestions experimentally?
3) Answer the question at the end of section 9.10.5.a.
4) The order of two reactions is given below:
i) 2A + B ![]() 2C.............
third order
2C.............
third order
ii) 2D ![]() 4E + F.............
first order
4E + F.............
first order
a) Write a possible rate expression for reaction i.
b) What is the probable order of reaction i with respect to A?
c) An experiment was performed on reaction ii. The initial concentration of D was 0.64moldm-3. Plot the relationship between [D] and time for this experiment given that the half life for the reaction is 5 seconds.
d) Reaction ii was repeated under different conditions. The initial concentration of A was 4moldm-3 and the initial rate was found to be 2.3 x 10-3moldm-3s-1. Calculate k for the reaction under these conditions, stating its units.
Unless otherwise stated, all materials in this web version of chapter 9 are © 2007 Adrian Faiers MA (Oxon) MCIPR

What 's the connection between a dozen eggs and
a garden mole?

Answer: Not a lot, really, but see Chapter 1